Soft Tissue Tumours
Clinical Leadership and Expertise1
Presented by Dr. Lalima Tiwari, an Oral Medicine Specialist and Clinical Senior Lecturer.
Learning Outcomes
This section provides comprehensive knowledge of soft tissue (mesenchymal) tumours, encompassing developmental, reactive, and neoplastic lesions.
Clinical Competencies for Developmental and Reactive Lesions2
Students should be able to describe the aetiology, pathogenesis, clinical and histopathologic features, diagnosis, and treatment of the following conditions:
- Vascular malformations
- Fibro-epithelial polyps
- Fibrous epulides
- Denture hyperplasia
- Inflammatory papillary hyperplasia
- Pyogenic granuloma
- Neoplastic lesions including Neurofibroma, neurilemoma, lipoma, hemangioma, rhabdomyosarcoma, fibrosarcoma, and Kaposi’s sarcoma.
Introduction to Mesenchymal Tumours3
Mesenchymal tumours originate from precursor cells derived from the mesoderm. These tumours can arise from various connective and supportive tissues, including:
- Bone
- Cartilage
- Blood vessels
- Adipose tissue
- Smooth muscle
- Fibroblasts
Clinical Objectives
The primary objective is to formulate appropriate diagnosis and treatment plans for mesenchymal tumours by identifying their clinical and histopathologic features.
Vascular Malformations
Definition
Vascular malformations are non-neoplastic developmental anomalies. Unlike true neoplasms, they do not contain hyperplastic cells but consist of progressively enlarging, aberrant, and ectatic vessels.
- Vascular malformations are named by the predominant vessel type:
- capillary
- lymphatic
- venous
- aterial
- Mixed
Aetiology
- Genetic mutations
- Molecular changes related to syndromes
- Trauma
- Blood flow and vascular wall resistance
Pathogenesis
- Do not contain hyperplastic cells
- progressive enlargement of aberrant and ectatic vessels composed of a particular vascular architecture
Clinical Features of Vascular Malformations
Vascular malformations present with a wide range of age manifestations, with 4-10% of newborn children presenting with congenital lesions often labeled as birthmarks. Clinical features vary according to the extension of the lesion, its location, and the displacement of neighboring tissue.
Capillary and Slow-Flow Malformations4
- Port wine stain: The most common vascular malformation. It can affect the skin, oral mucosa, and sclera, appearing either in isolation or as part of a systemic disease.
- Slow-flow malformations: Typically detected at birth.
- Color varies depending on depth, ranging from normal skin color to deep purple.
- Must be distinguished from haemangioma, which exhibits continuous growth.
High-Flow Vascular Malformations
- Often manifest in the 3rd decade of life.
- Typically present with a bluish color.
- Often manifest significant changes during puberty.
- Arteriovenous malformation (AVM): Characterized by swelling, pain, and bleeding. These are pulsatile and not fluctuant.
Mixed and Intraosseous Presentations
- Mixed lesions: Behave as high-flow due to arterial supply; should be distinguished from Hereditary Hemorrhagic Telangiectasia (HHT).
- Intraosseous lesions: Present with poorly defined borders or well-circumscribed halos.
- Radiographic appearance ranges from unilocular or multilocular to unicystic or honeycomb patterns.
- May cause bone expansion.
Classification by Blood Flow Pattern5
The following table categorizes the most common vascular malformations of the head and neck based on their blood flow characteristics:
| Slow Flow | High Flow |
|---|---|
| Sturge-Weber syndrome | Arteriovenous malformations |
| Venous malformations | Arteriovenous fistula |
| Capillary malformations | Capillary arteriovenous malformation |
| Capillary venous malformation | Capillary lymphatic venous arteriovenous malformation |
| Capillary lymphatic malformation | |
| Lymphatic venous malformation | |
| Capillary lymphatic venous malformation |
Histopathology of Vascular Malformations
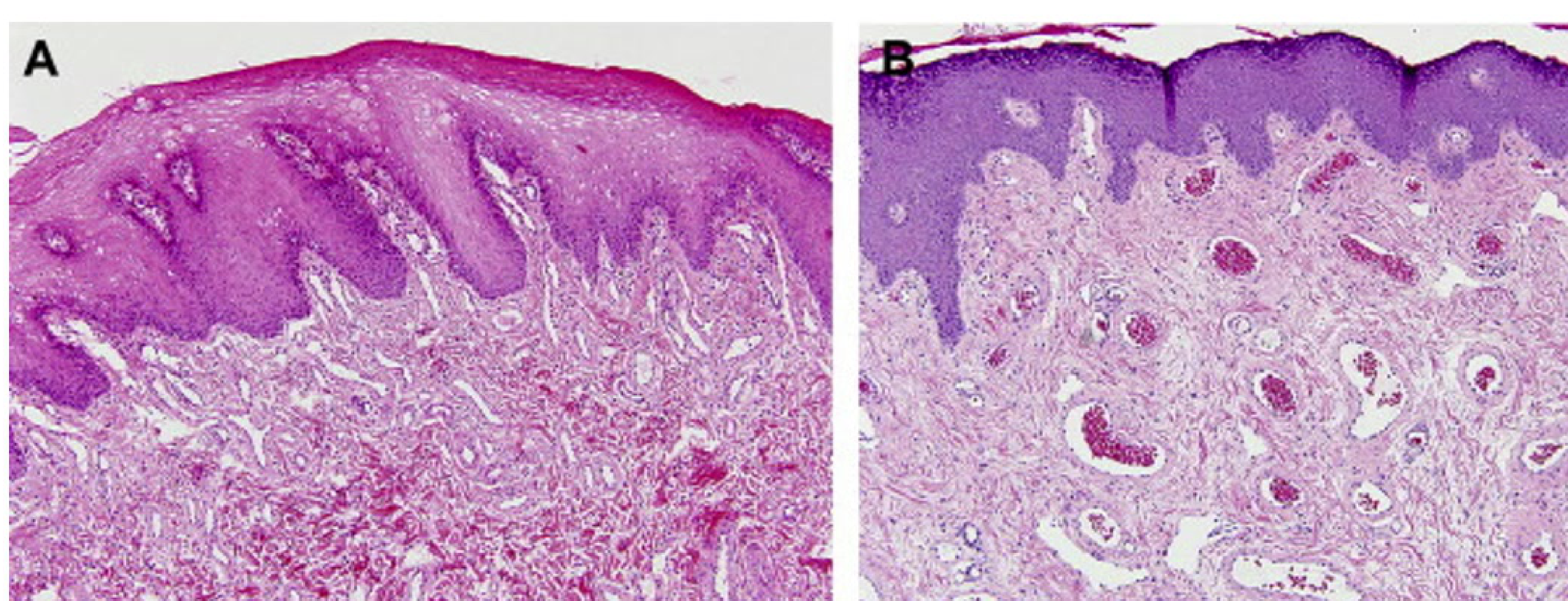
Capillary Malformation (CM) Histology6
- Facial CM with lip hypertrophy (4-year-old): Shows an excessive number of thin-walled venule-like channels with narrow lumens.
- Facial CM with lip hypertrophy (17-year-old): Characterized by an increased number of enlarged vein-like channels with both thin and thick, mostly fibrous walls. There is an increase in intervascular fibrous tissue.
- Adults often show haphazardly arranged ectatic capillaries and venules.
- Features include flat endothelial cells, thin collagenous walls, and layers of pericytes.
- Facial CM in Sturge-Weber syndrome (35-year-old): Nasal skin shows a nodular cluster of large, abnormal vein-like channels. Histology reveals fibrosis, follicular dilatation, and keratin plugging.
 |  |
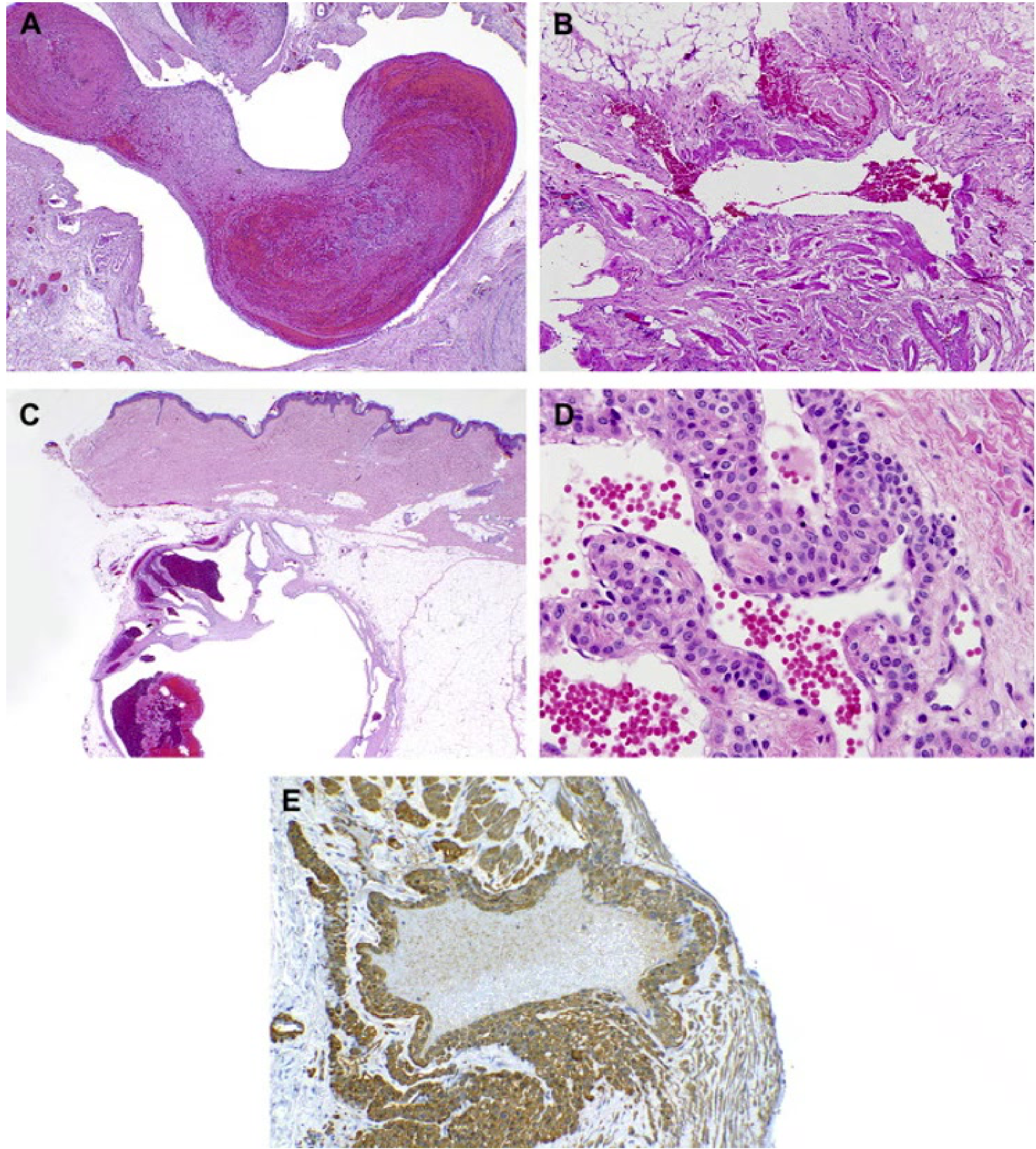
Venous (VM) and Glomuvenous Malformation (GVM) Histology7
- A. Venous Malformation (VM): Exhibits malformed venous channels often containing organizing thrombi. B. The venous walls are irregularly muscularized and may focally lack muscle.
- C. Glomuvenous Malformation (GVM):
- C. Deep dermal/subcutaneous lesions often present with organizing thrombi.
- D. Vascular channels feature cuboidal glomus cells that replace the standard smooth muscle.
- E. Glomus cells can be specifically highlighted using smooth muscle actin immunostaining.
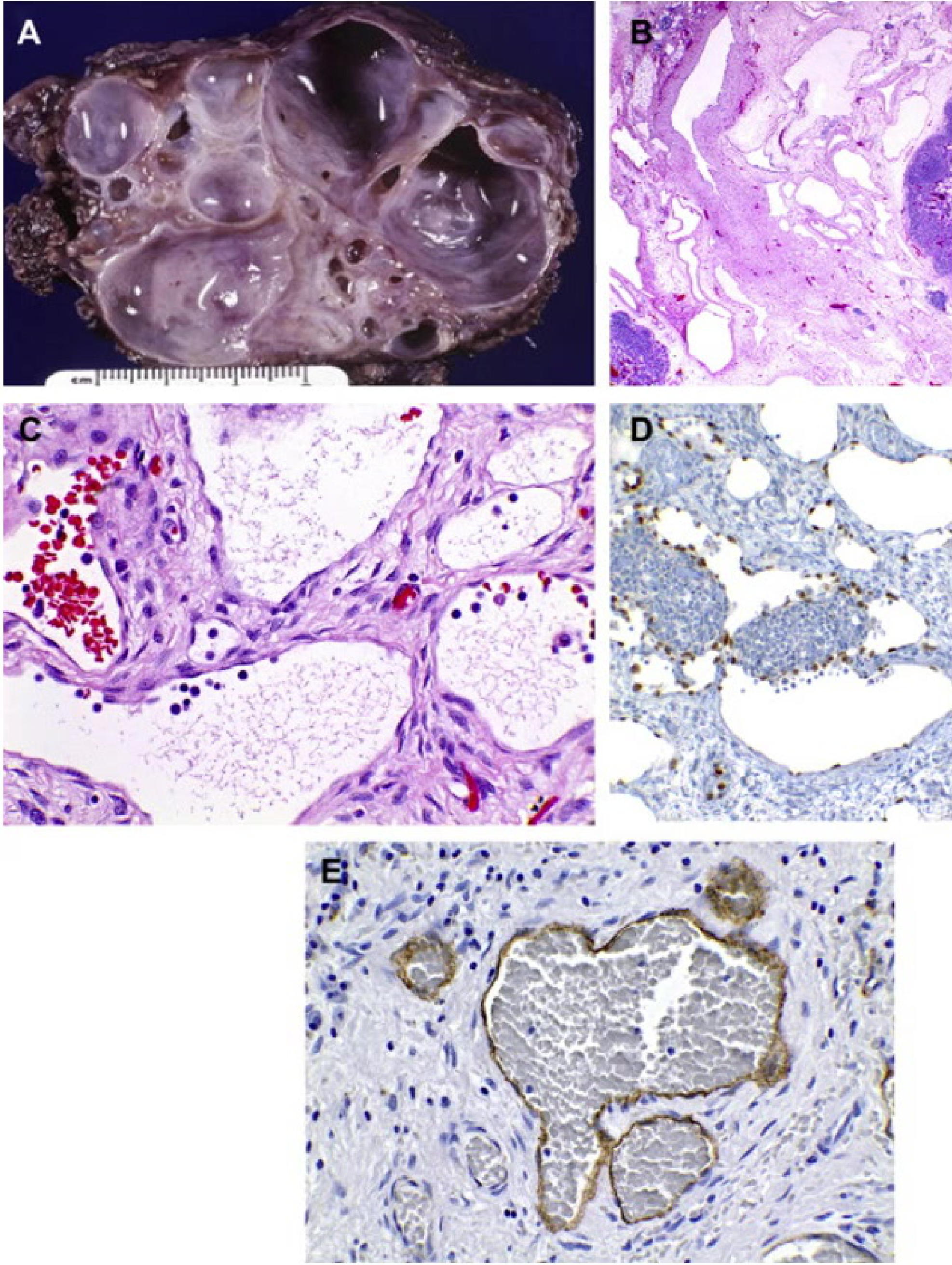
Lymphatic Malformation (LM) Histology8
- A. Macrocystic LM: Characterized by cyst-like channels measuring 1 cm or greater.
- B. Microcystic LM: Features vascular channels less than 1 cm in size.
- C. Channel Characteristics: Small, thin-walled channels in LM possess thin endothelial cells without an apparent muscular wall. Lumens typically contain protein and lymphocytes.
- The lesions are often described as "sponge-like".
- Enlargement is primarily due to lymph fluid distension.
- Immunohistochemistry:
- D. PROX-1: Highlights lymphatic endothelium with a brown nuclear stain.
- E. D240: Highlights lymphatic endothelium with a brown cytoplasmic stain.
Arteriovenous Malformation (AVM) and PTEN Hamartoma (PHOST) Histology9
- Arteriovenous Malformation (AVM):
- A. Shows malformed arteries and veins.
- B. Verhoeff-van Gieson (VVG) stain highlights a disrupted internal elastic lamina in arteries with a transition to an indeterminate elastic pattern.
- C. Proliferative components may show small channels with plump endothelium and pericytes.
- PTEN Hamartoma of Soft Tissue (PHOST):
- D. Intramuscular PHOST: Features large nodules composed of large veins with irregular lumens and focally hypermuscularized walls, surrounded by prominent arteries.
- E. Tissue Composition: Contains dense fibrous tissue, small myxoid vascular nodules, and lymphoid clusters.
- F. Vascular Clusters: Some clusters are composed of very thin-walled abnormal veins resembling pulmonary alveoli; tortuous arteries may show transmural muscular hyperplasia and small lumens.
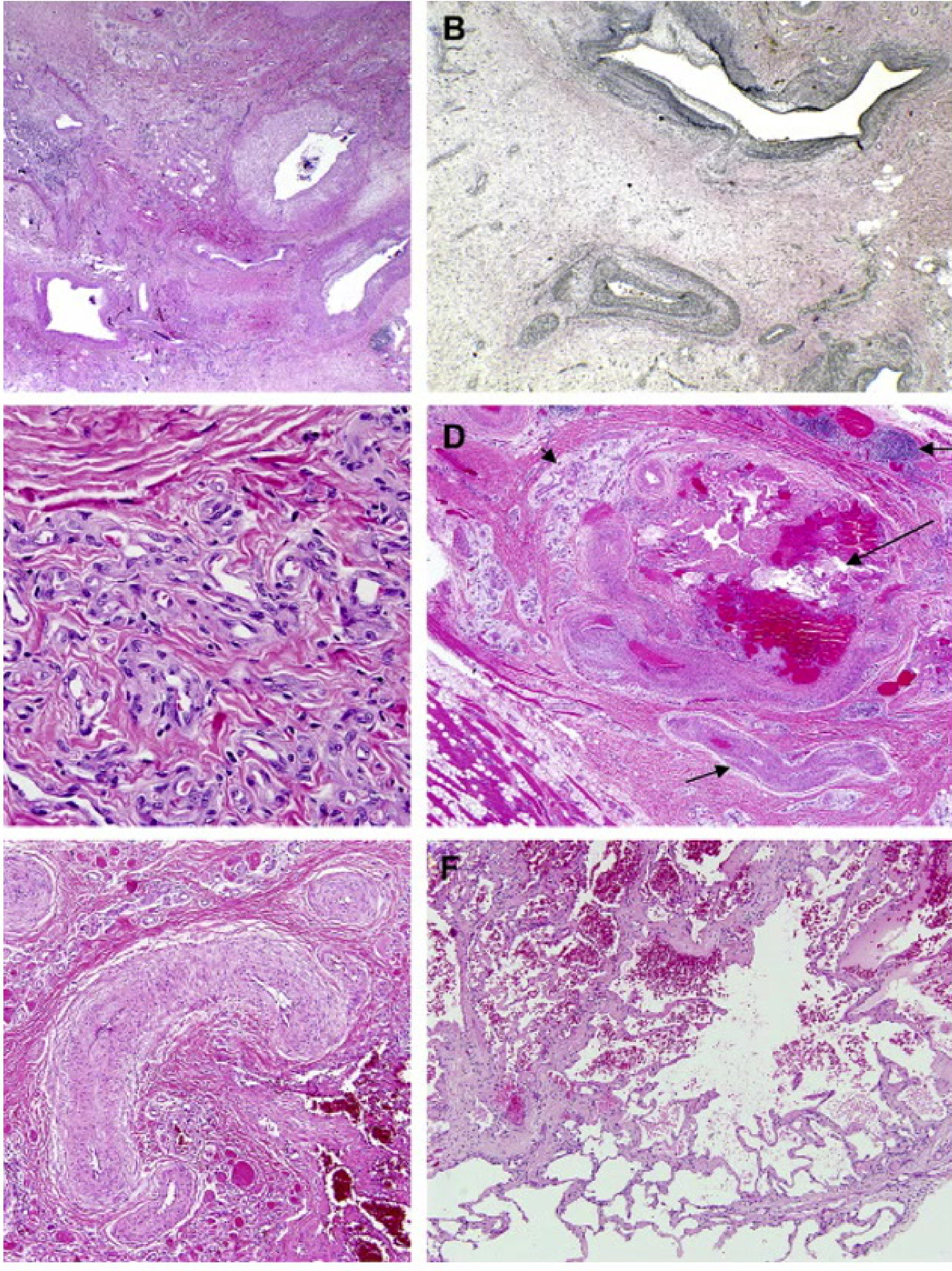
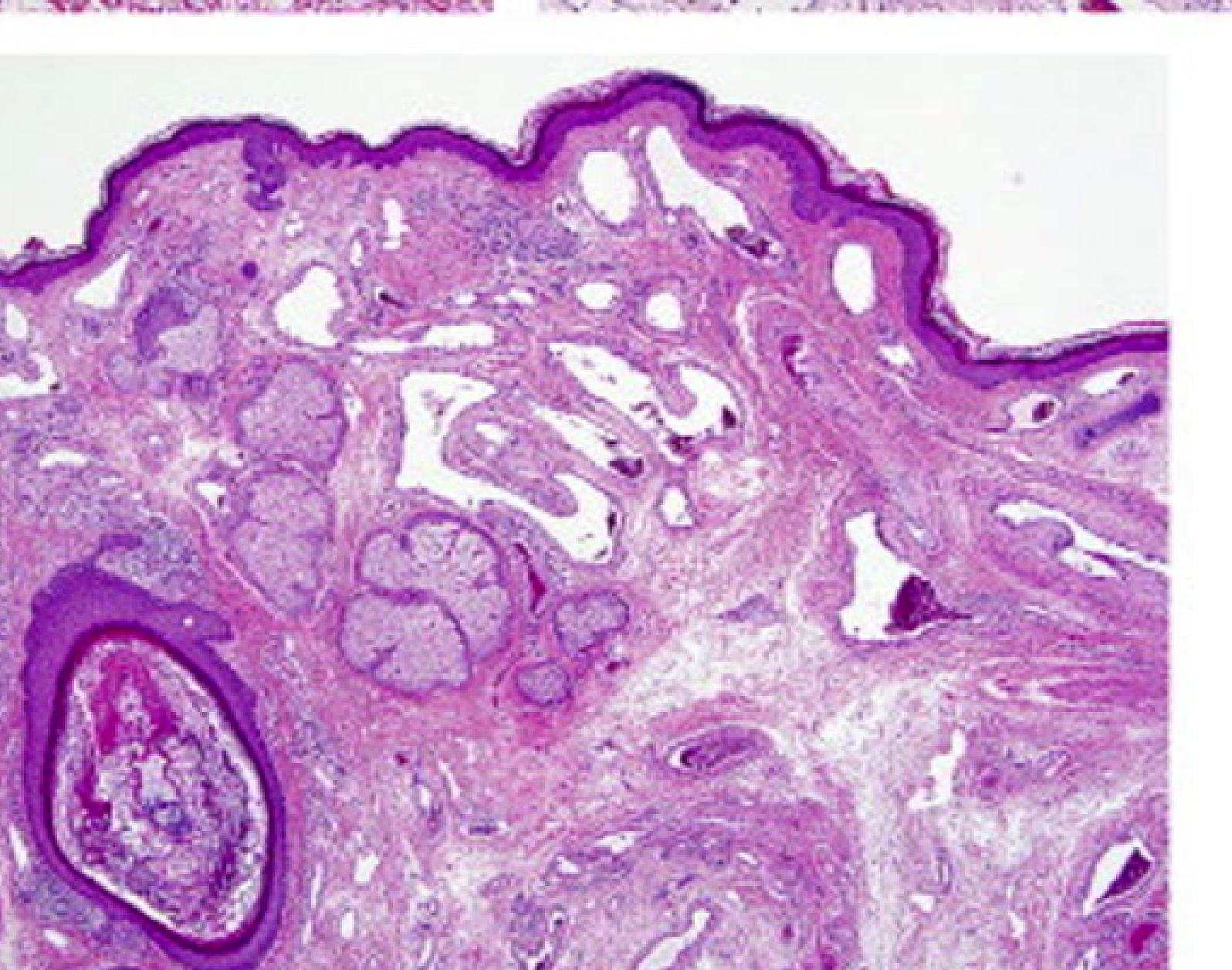
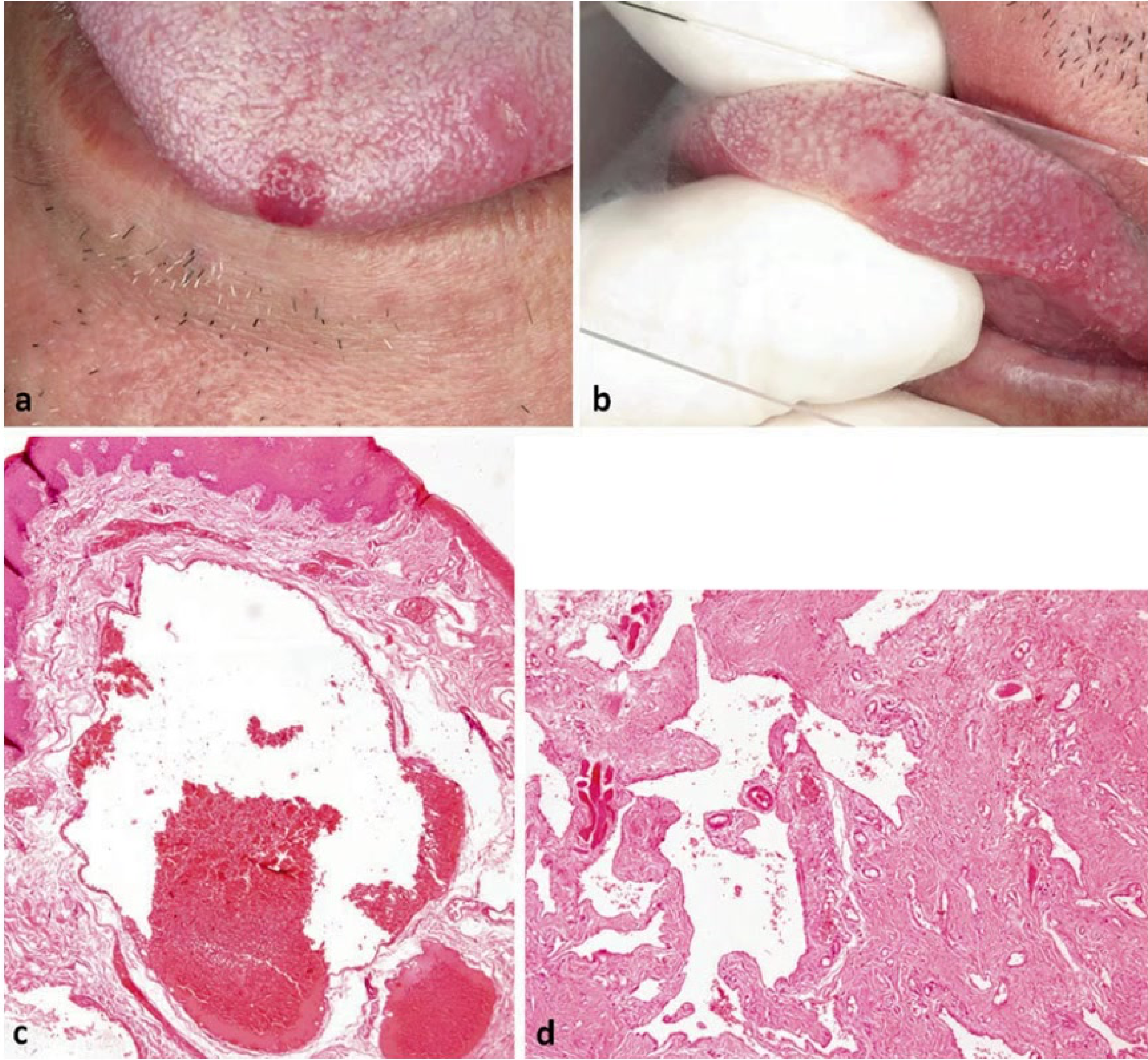
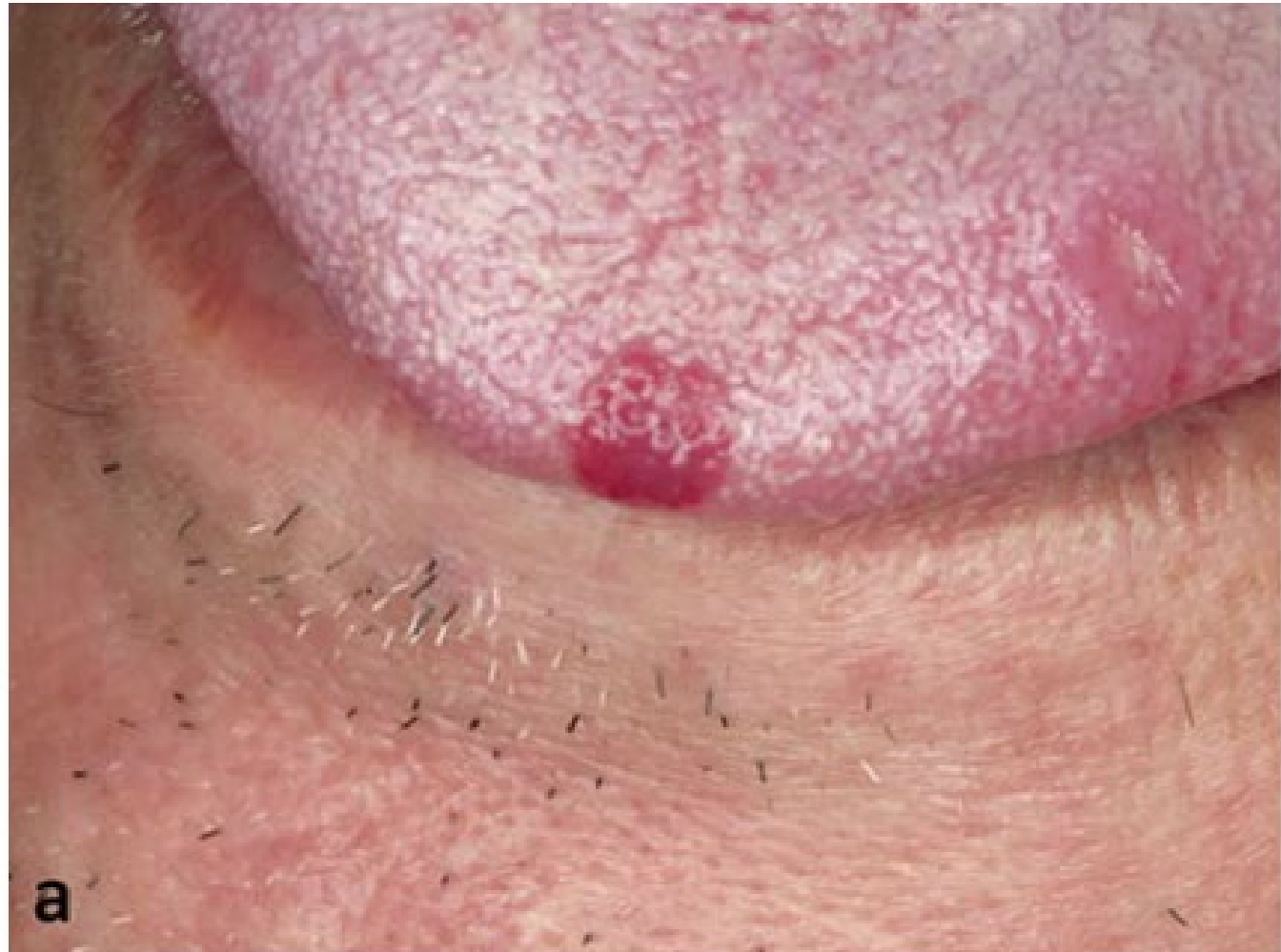
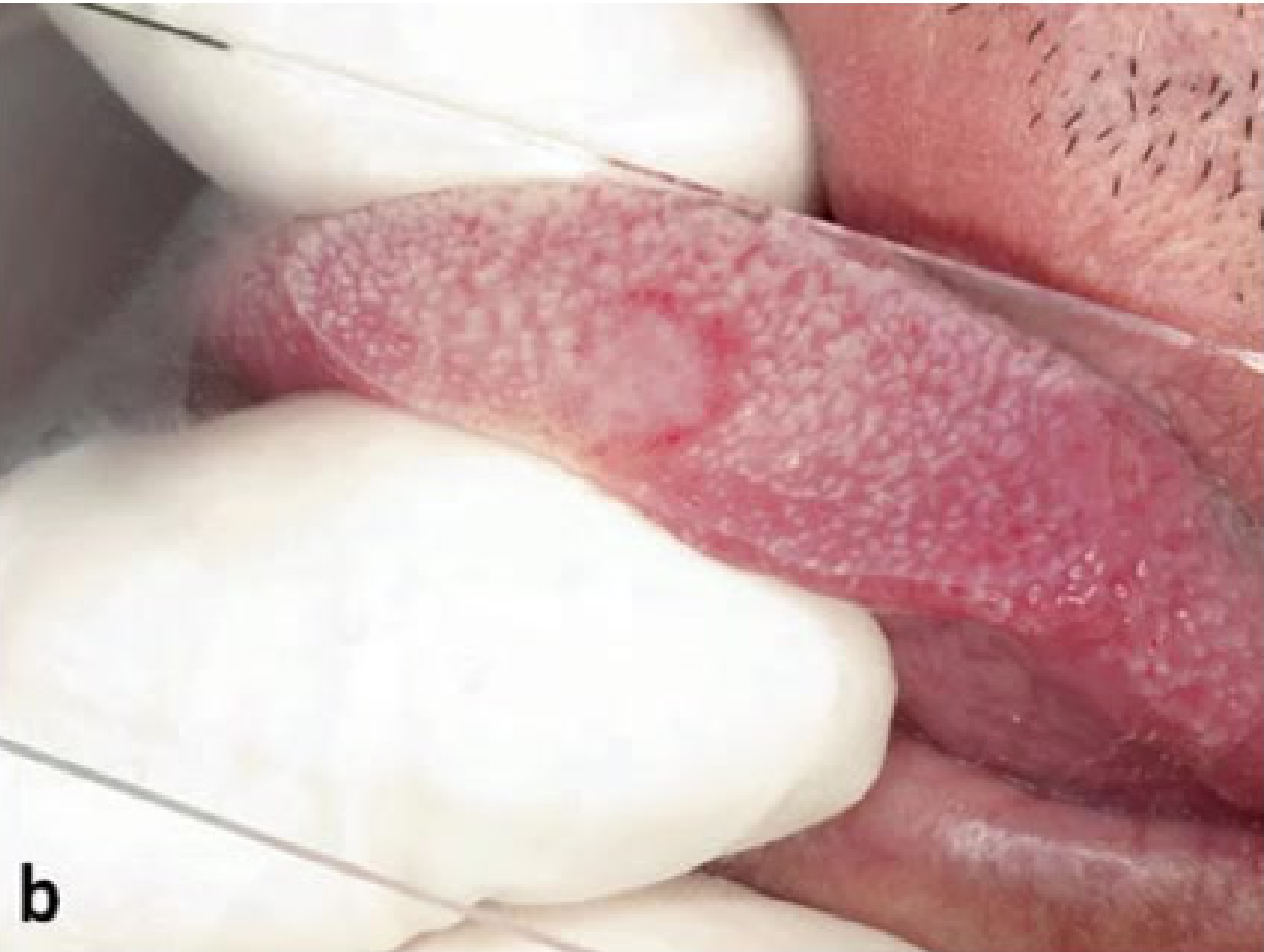
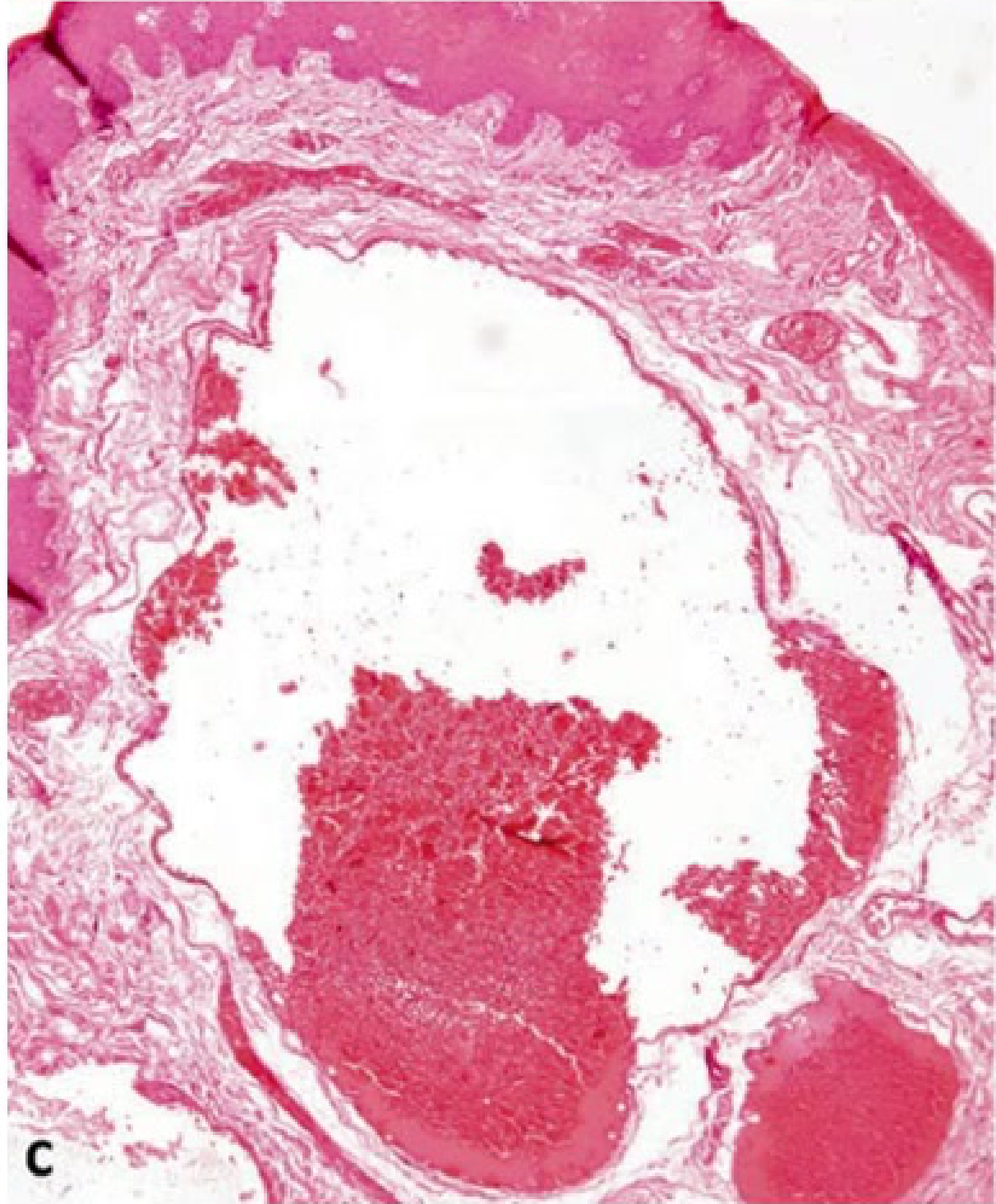
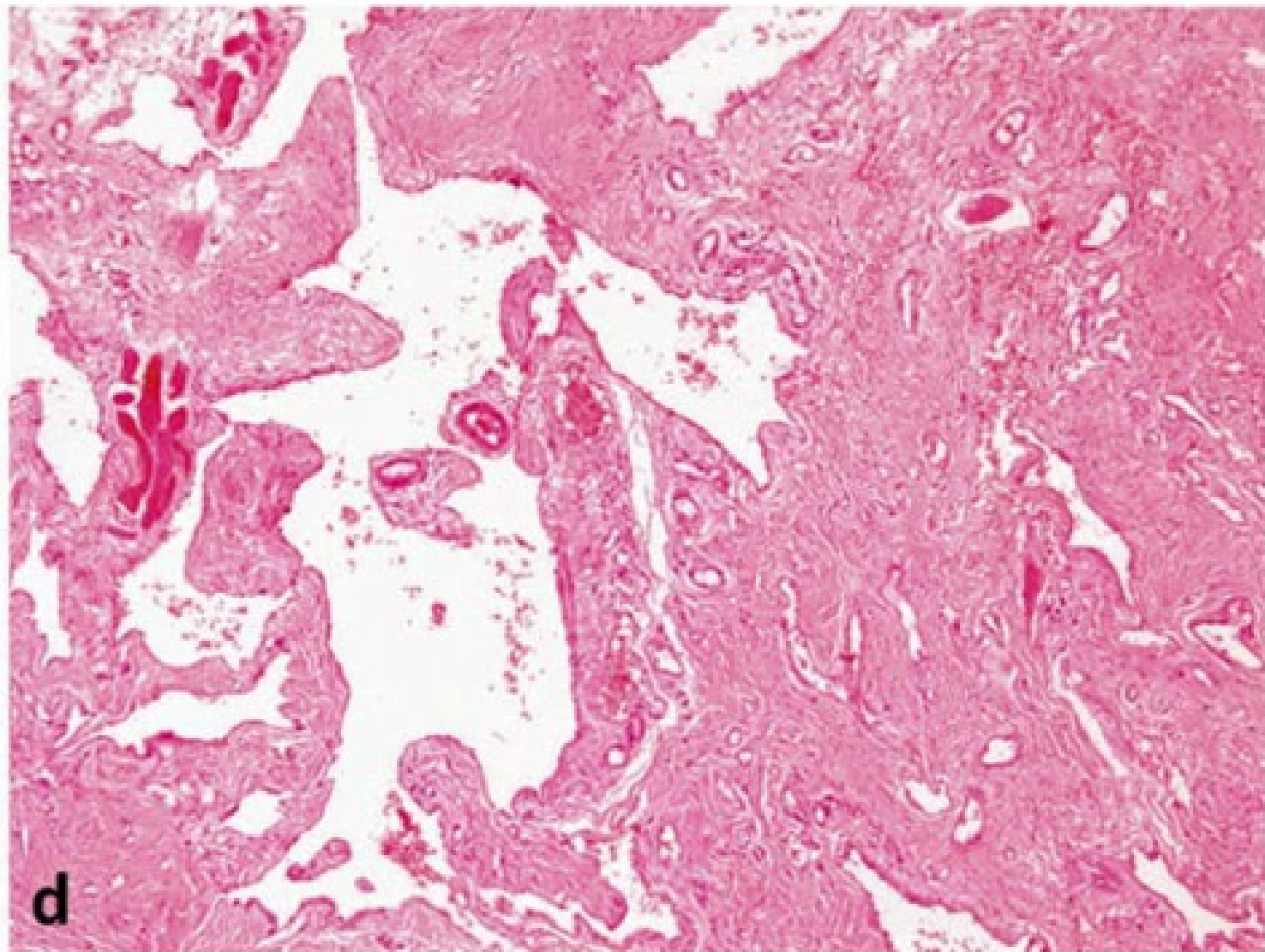
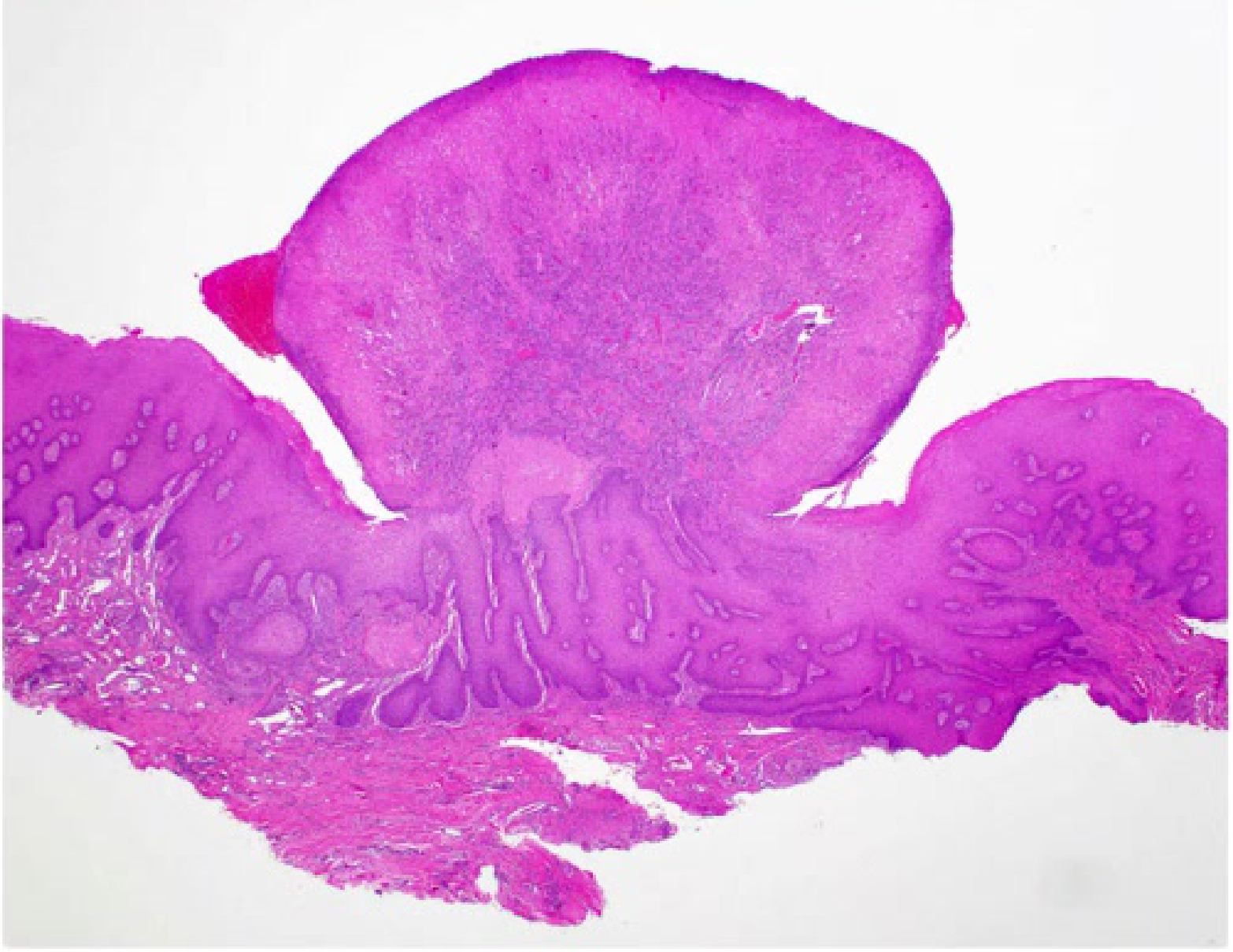
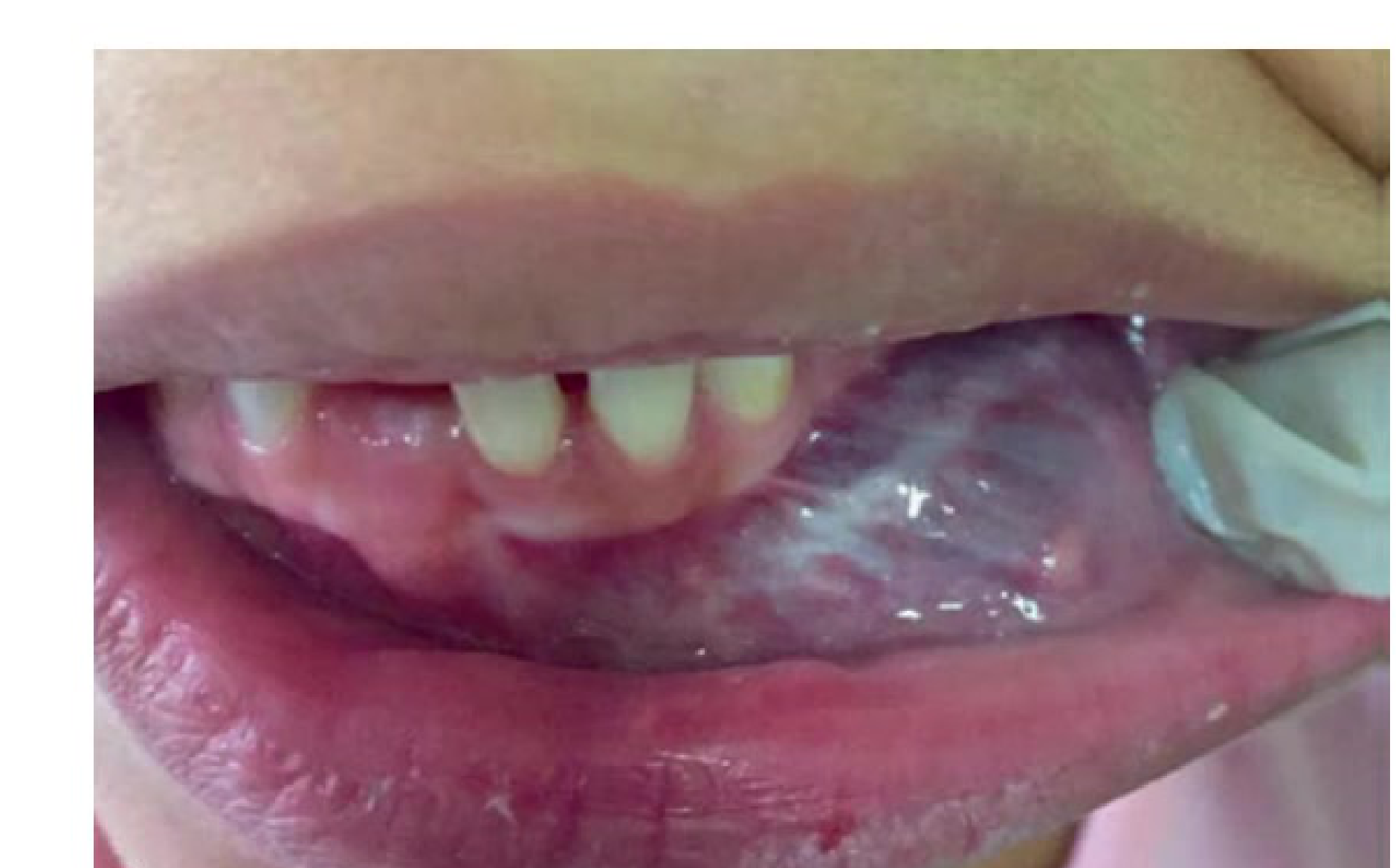
Clinical and Microscopic Presentation: Tongue Apex10
- Clinical Presentation: A vascular malformation at the apex of the tongue appearing as a red patch.
- Diagnostic Procedure: Diascopy (blanching procedure) reveals a whitish appearance upon compression, confirming the vascular nature
- A glass slide test (droscopy) will show complete blanching, followed by a refill once pressure is released..
- Microscopic Findings: Low power magnification of a venous-capillary malformation reveals multiple blood vessels of variable width.
 |  |  |
 |  |
Diagnosis and Treatment of Vascular Malformations
Accurate diagnosis is essential for appropriate treatment planning.
Diagnostic Modalities11
- Microscopic Evaluation: Provides the most detailed impression of vascular alterations. It is critical for differentiating malformations from haemangiomas. A key histological marker is the consistent presence of nerve bundles within vascular malformations.
- Imaging Tools:
- MRI: Considered the best imaging tool for both diagnosis and treatment planning - T2-weighted MRI reveals hyper-intense irregular lesions with "flow voids"..
- Arteriography: Magnetic resonance arteriograms (MRA) and computed tomograph arteriograms (CTA) provide excellent visualization of Arteriovenous Malformations (AVM).
- Hyper-contrast CT: Utilized for further examination of the lesion’s extent.
High-Flow Arteriovenous Malformation Case Study12
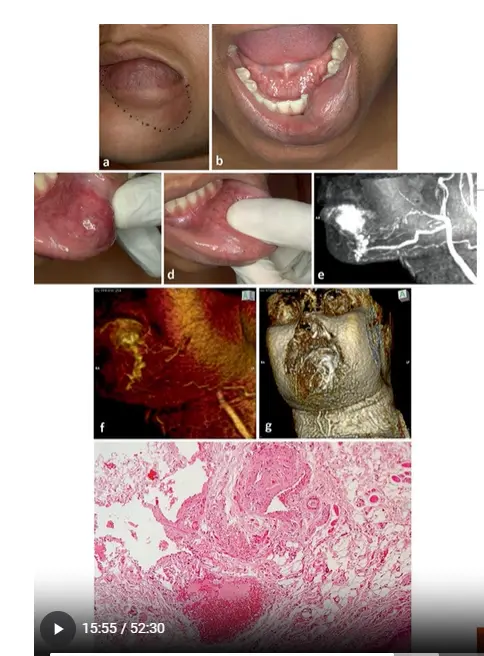
- Clinical Presentation: A pinkish, non-fluctuating nodule extending from the midline of the lower lip to the labial commissure.
- Clinical Examination: Compression reveals pulsating behavior, indicating a high-flow pattern.
- Imaging (MRA): Reveals an arterial origin from the facial artery and an arteriovenous fistula forming a plexiform arrangement with dilated vessels and an effluent mental vein.
- Microscopic Aspect:
- Demonstrates a close relationship between thick-walled arteries with narrow lumens and thin-walled veins with dilated, blood-filled lumens.
- The lesion infiltrates surrounding tissues, including adipose tissue, striated skeletal muscle, and neural fibers.
Management of vascular malformations usually requires a multi-disciplinary center approach.
Treatment Modalities13
- Conservative Measures: Generally only effective for venous malformations
- Includes head-of-bed elevation and compression..
- Electrocautery: Used for targeted tissue destruction.
- Laser Therapy: Utilized for superficial or specific vascular components
- Sclerotherapy is also a common interventional option..
- Combined Therapy: Embolotherapy combined with surgical excision is frequently required, particularly for Arteriovenous Malformations (AVM).
Reactive and Developmental Fibrous Lesions
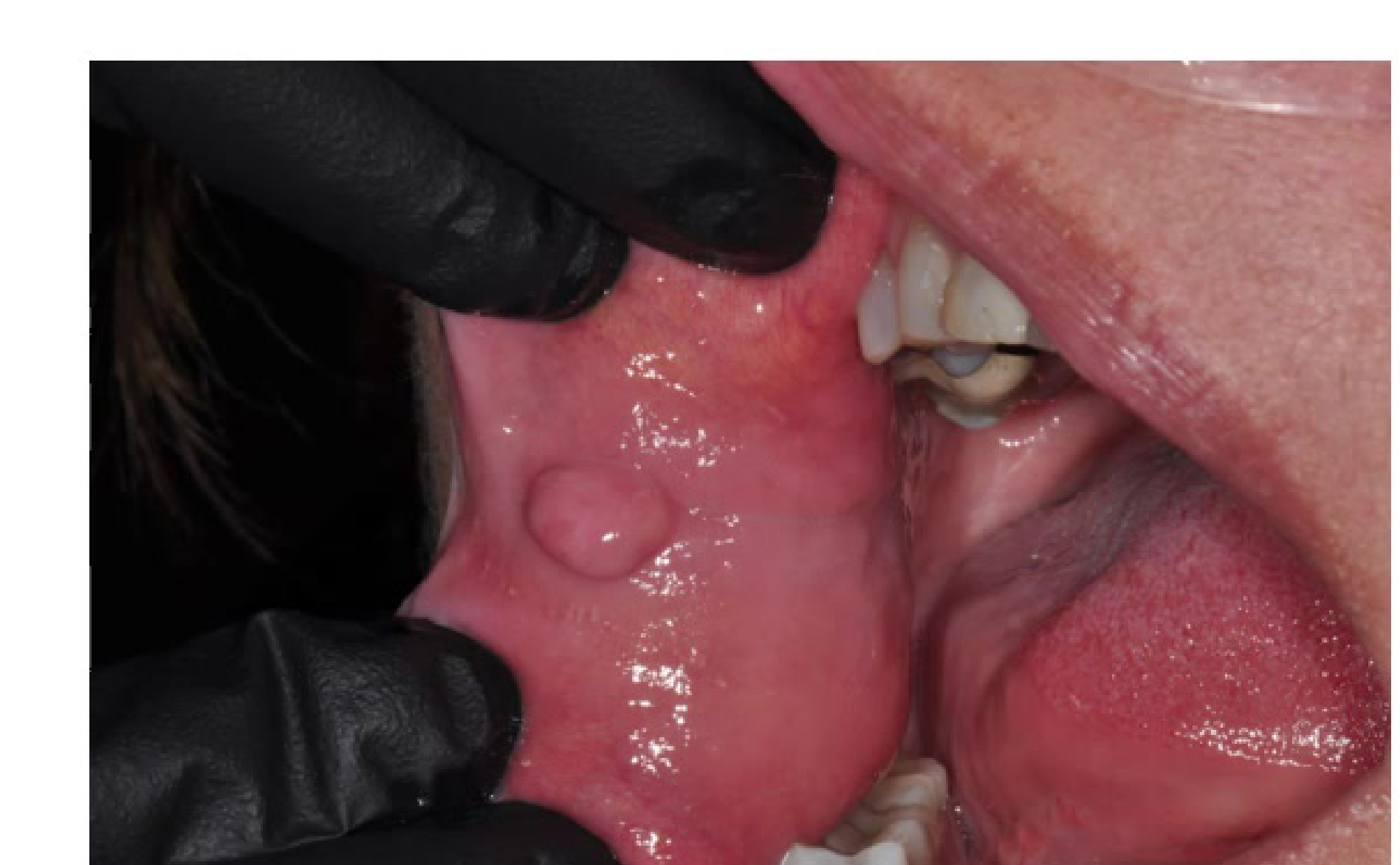
Fibroepithelial Polyp
- Fibrous nodule
- Aetiology: Induced by recurrent local irritants of oral cavity – bite trauma, denture irritation, food impaction, poor oral hygiene
- Pathophysiology: Reactive proliferation rather than true neoplasm
Clinical Features14
- Smooth, round, exophytic nodule
- Pedunculated or sessile with normal overlying epithelium
- Can be ulcerated, or demonstrate thickened white surface (hyperkeratosis)
- Asymptomatic generally
- 1-2 cm in diameter
- Labial mucosa, tongue, palate
Histopathology15
- Nonencapsulated nodular mass
- Mass composed of fibrous connective tissue with collagen bundles interspersed with fibroblasts, blood vessels and scattered chronic inflammatory cells
- Overlying surface of squamous epithelium
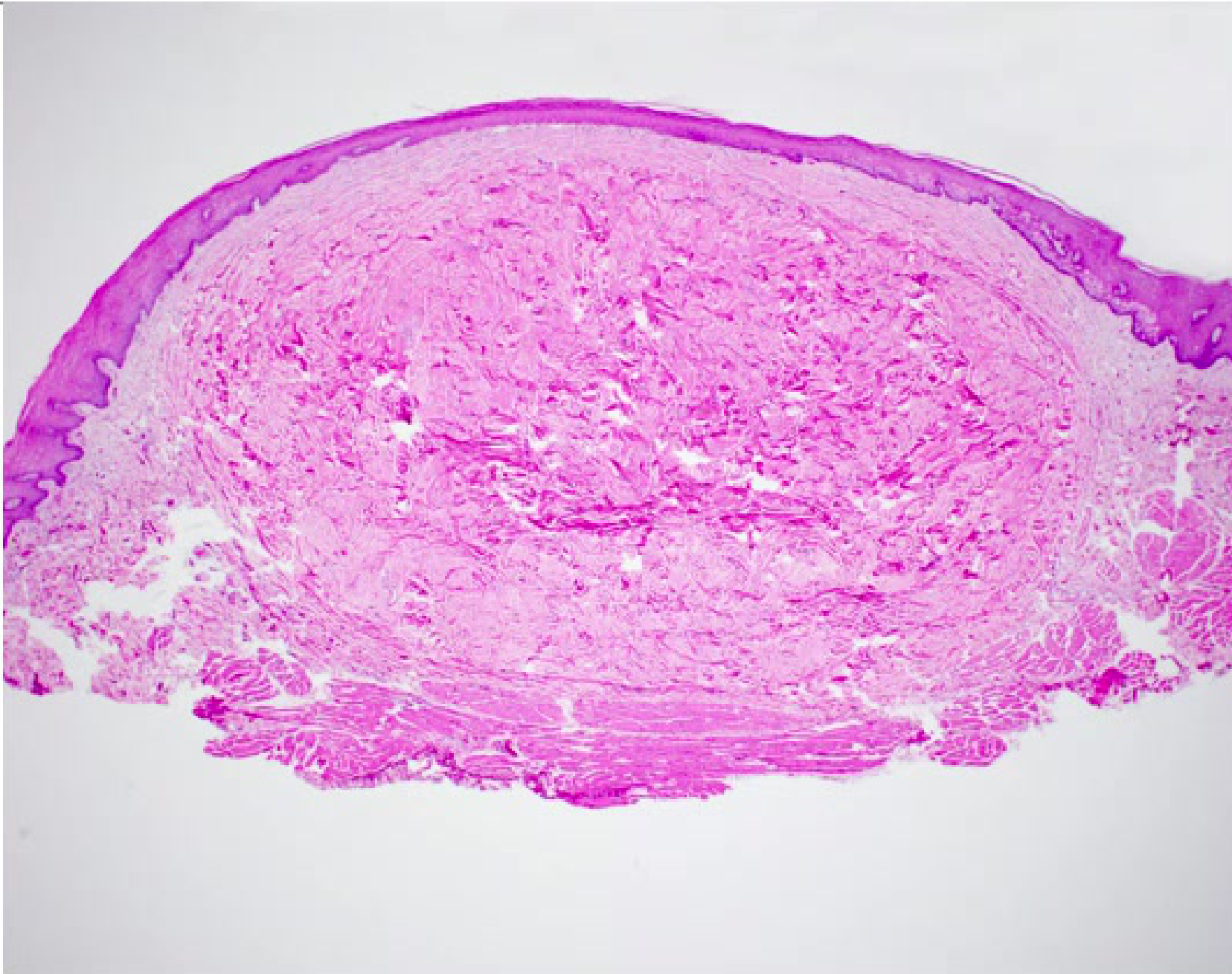
Diagnosis
- Definitive diagnosis made upon histopathological examination
- Definitive diagnosis via excision biopsy.
- Highly suspected based on clinical features
Treatment
- Surgical excision with removal of local irritants
- Removal of the local irritant (e.g., sharp tooth or ill-fitting denture) is essential to prevent recurrence.
Fibrous Epulides
Fibrous lump on the gingiva representing a localised hyperplastic fibrous gingival mass formed as a response to chronic irritation.
Clinical Presentation16
- Smooth pink nodule on marginal/attached gingiva
- Often around inflamed gingiva
- Often occurs as a response to chronic irritation from calculus or poor restoration margins.
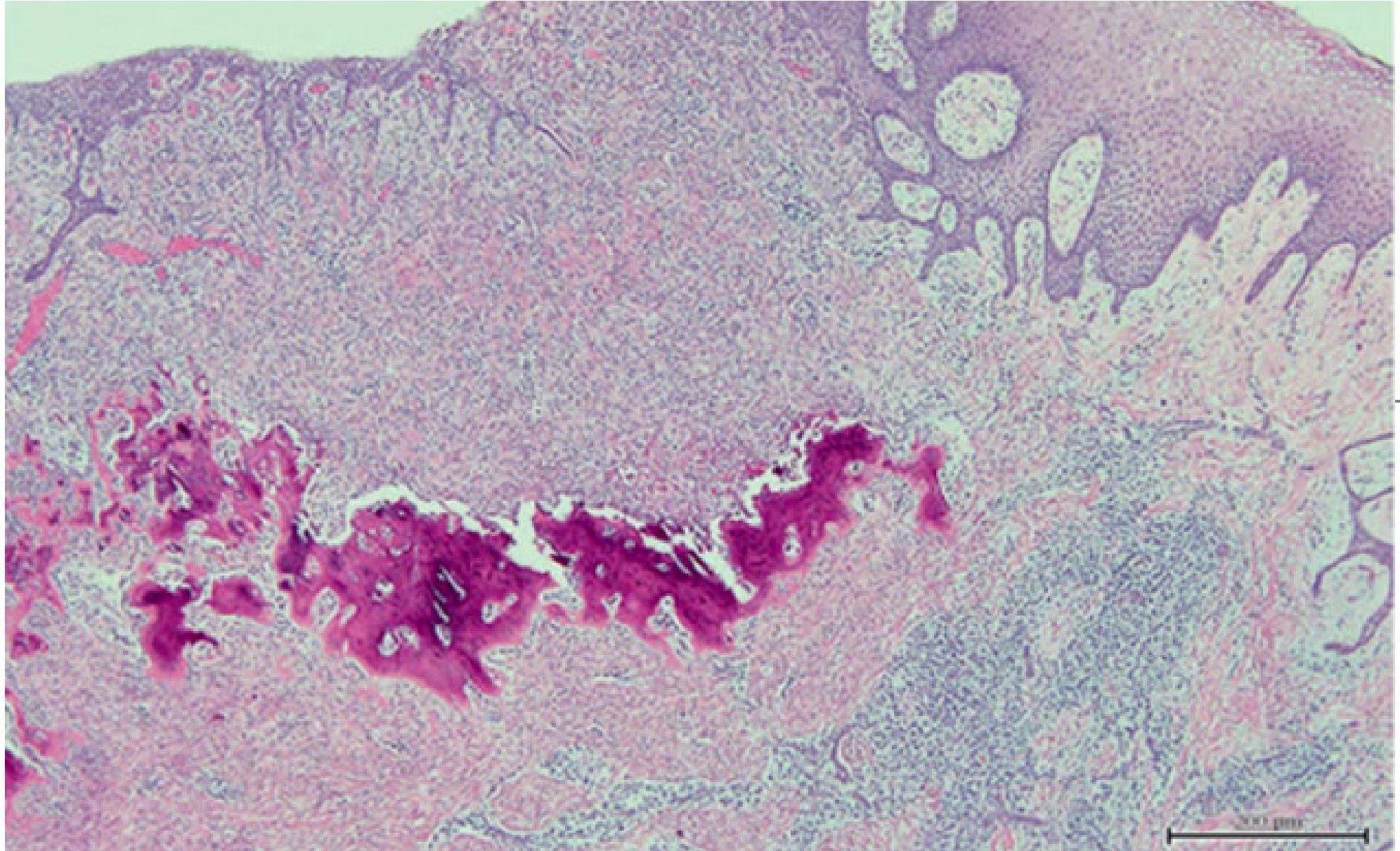 | 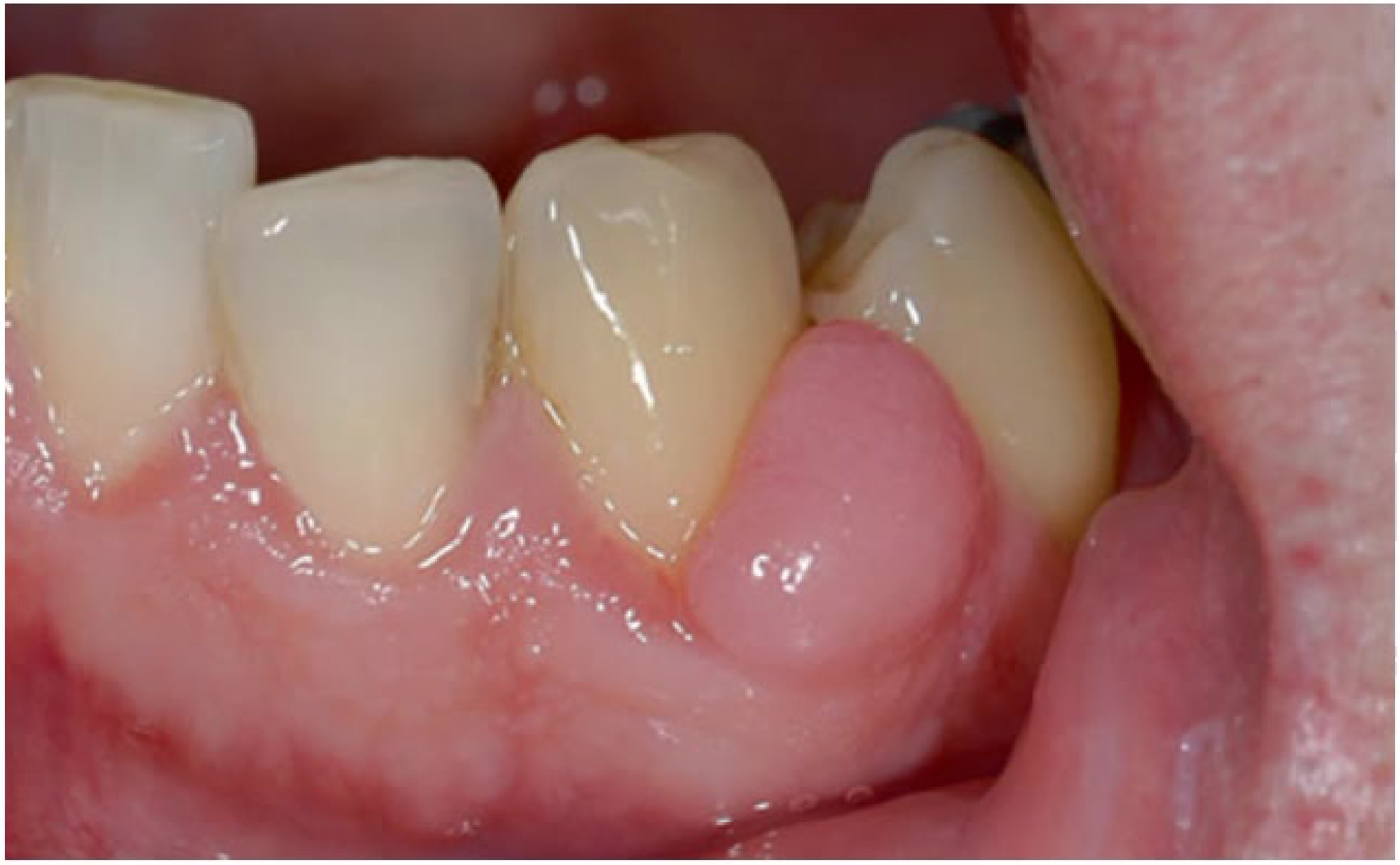 |
Histopathological Features
- Mature fibrous tissue
- Minimal inflammation
- Mineralisation commonly seen – dystrophic calcification
Diagnosis and Management
- Histopathological evaluation needed to confirm diagnosis
- Surgical excision to periosteum, scale and clean, with removal of local irritants
- Requires scaling and root planing of the area.
- Recurrence can occur if irritants not removed
Denture Hyperplasia
Also known as denture fibroma or flabby ridge, this is an inflammatory hyperplastic reaction caused by chronic irritation, usually due to ill-fitting dentures.
Terminology
Also known as Epulis Fissuratum.
Clinical Features17
- Sessile or pedunculated, pink nodules
- Slow growing
- Located on alveolar ridges or denture bearing areas, usually seen under dentures
- Commonly found in the vestibular sulcus.
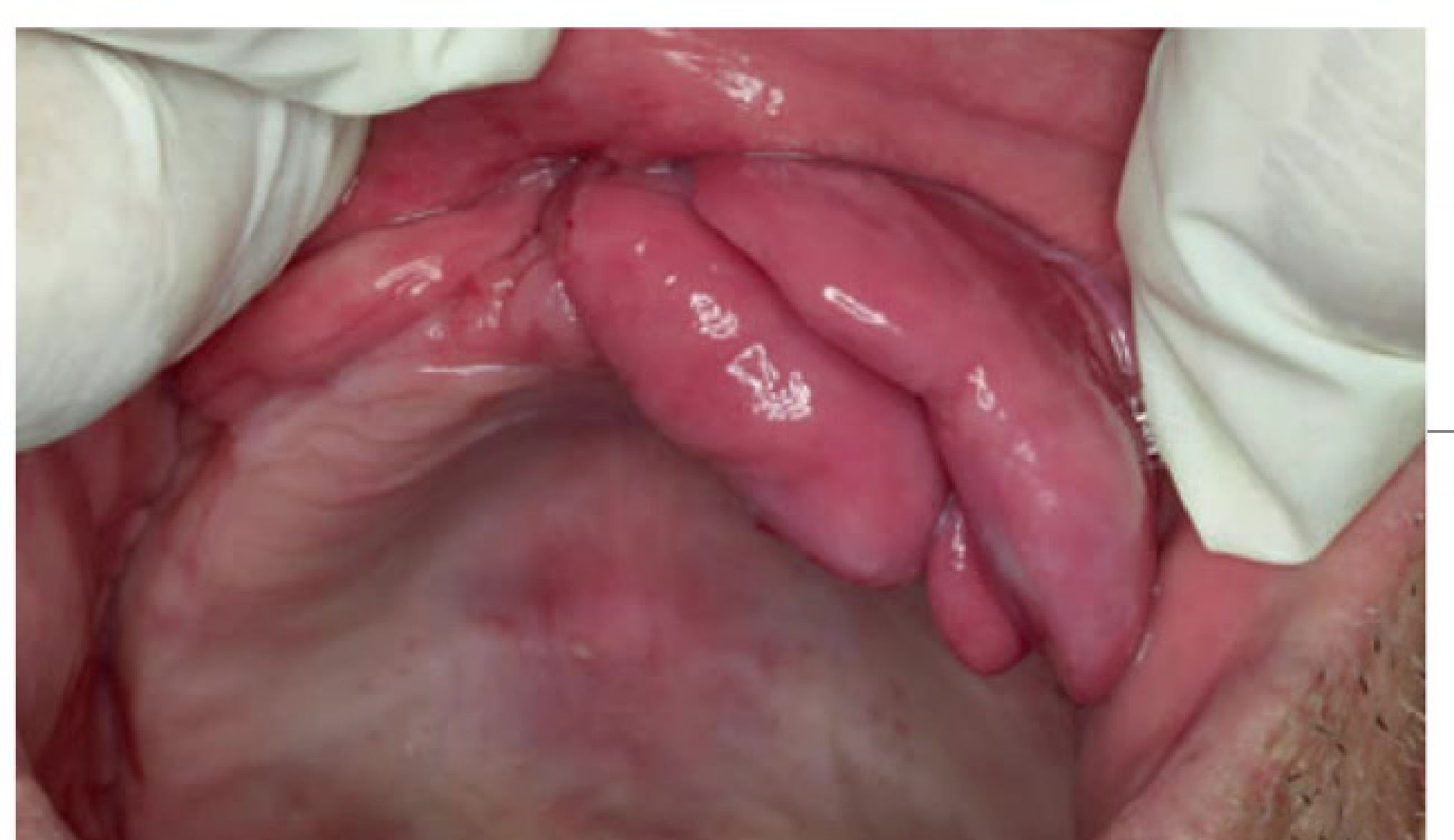 | 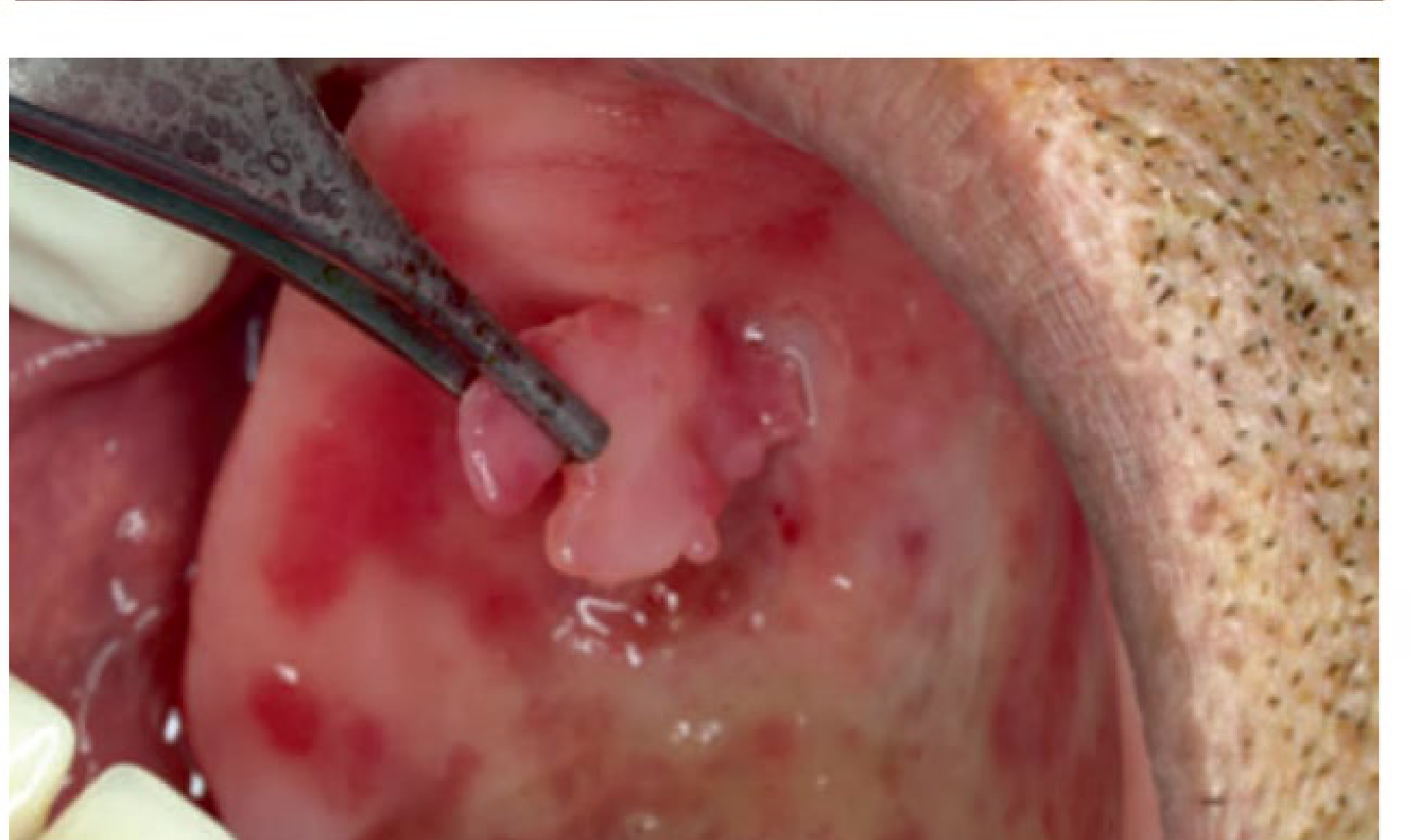 |
Histopathology
- Epithelial hyperplasia, acanthosis, pseudo-epitheliomatous hyperplasia
Diagnosis
- Based on clinical features and histopathological assessment
Management
- Local surgical excision
- New, well-fitting dentures
- Denture hygiene
Inflammatory Papillary Hyperplasia
Benign soft tissue lesion often associated with use of removable upper dentures; pathogenesis is unclear.
Aetiology18
- Ill-fitting dentures, continuous day and night denture use, poor oral hygiene, sensitivity to denture liners, tobacco
- Often associated with colonization of Candida caused by poor oral hygiene
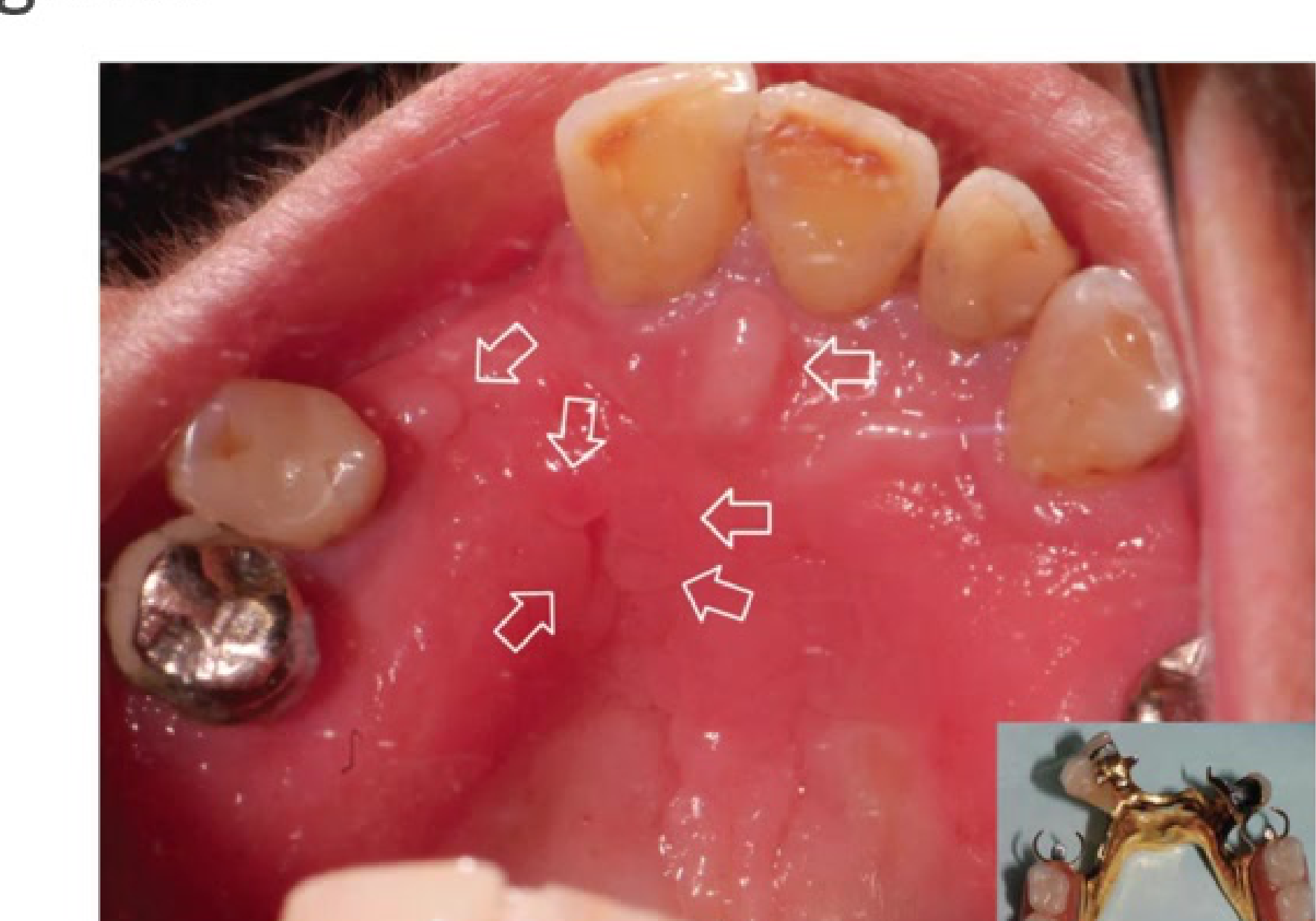
Clinical Features
- Growth of one or more nodular lesions
- 2mm or less in size
- Almost exclusively involves the hard palate
- Mostly asymptomatic
- Colour of mucosa may vary from pink to red
- Often presents with a "cobblestone" appearance.
Histopathology19
- Papillary projections covered by stratified squamous epithelium with or without chronic inflammation
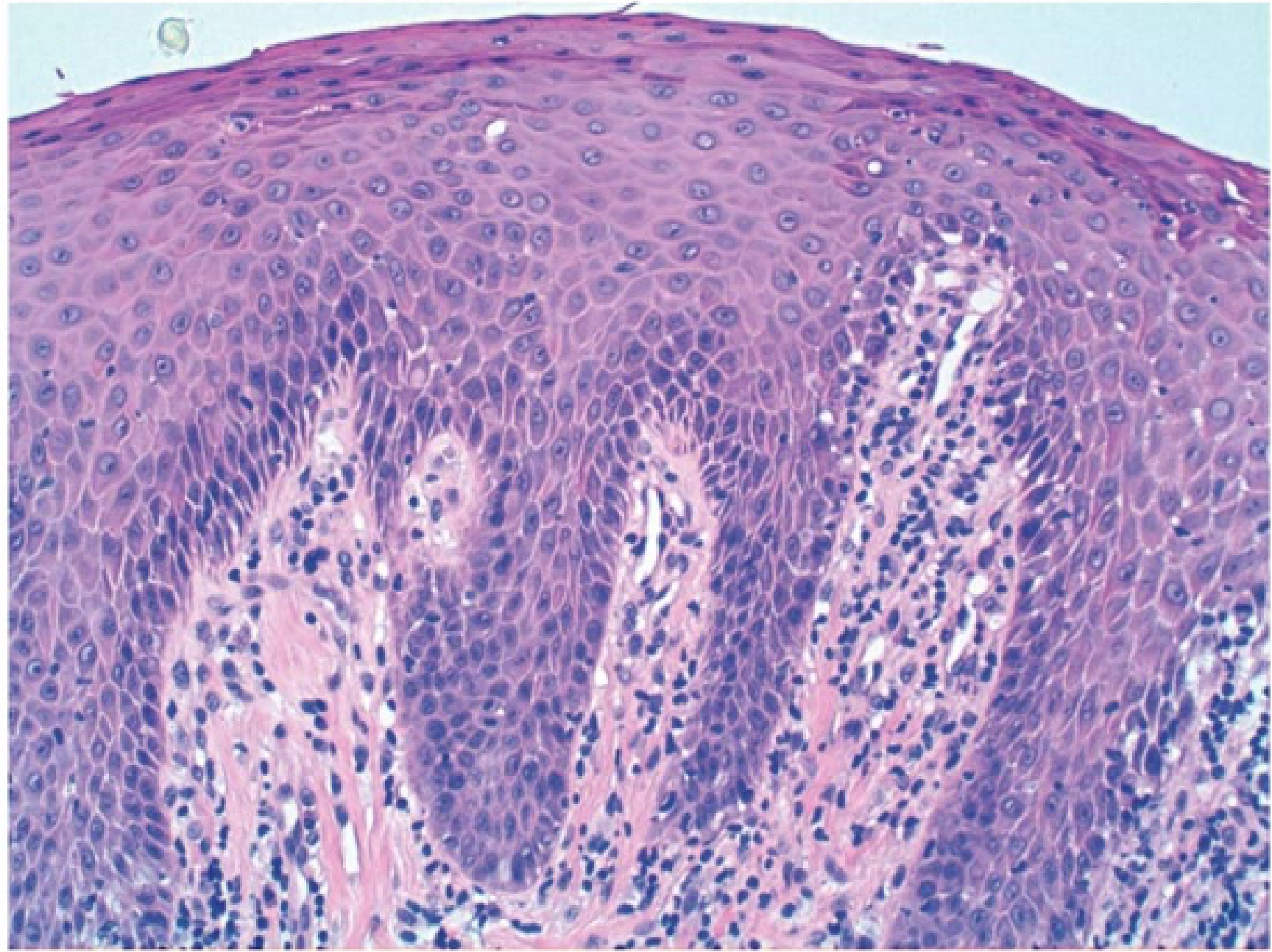
Diagnosis
- Clinical examination and histopathological evaluation
Treatment
Treatment depends on severity and may include:
- Laser
- Electrosurgery
- Cryotherapy
- Small localised lesions: 0.12% chlorhexidine mouthrinse, antifungal gels, local excision
- Antifungal therapy and improved hygiene are primary conservative treatments.
Pyogenic Granuloma
Benign soft tissue lesion.
Aetiology20
- History of trauma
- Response of tissues to minor trauma and/or chronic irritation
- Associated with poor oral hygiene, chronic trauma, pregnancy, medications
Pathogenesis
- Largely unknown, although in pregnancy, estrogen enhances vascular endothelial growth factor (VEGF) production in macrophages, likely contributing to development.
- Progesterone in pregnancy may function as an immunosuppressant in gingiva, preventing an acute inflammatory reaction against oral bacteria and resulting in proliferative gingival inflammation.
- Increased estrogen and progesterone in pregnancy increases concentrations of Prevotella intermedia in the subgingival biofilm, decreases the host response, and increases vascular permeability and fluid infiltration.
Clinical Features21
- Wide range of ages affected
- Gingiva most common site; can occur extra orally
- Soft, painless, deep red to reddish-purple in colour
- Bleeds easily upon provocation.
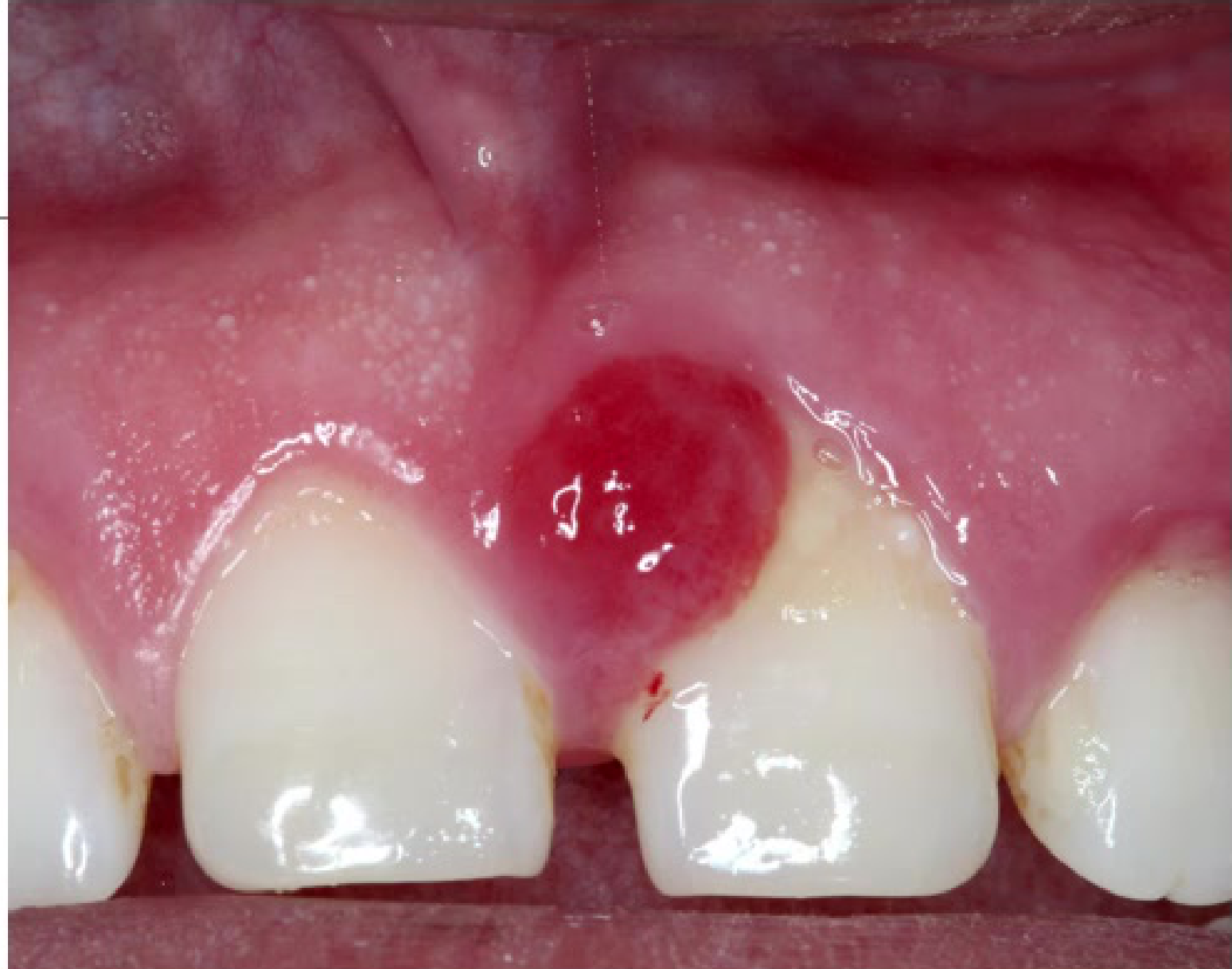 |  |
Histopathology
- Highly vascularized proliferation of granulation tissue
- Often demonstrates surface ulceration and a subacute inflammatory cell infiltrate comprised of neutrophils, lymphocytes and plasma cells
- May demonstrate a lobular arrangement of capillary vessels and proliferating endothelial cells delineated by fibrous septae (termed lobular capillary hemangioma)
- May be pedunculated
- May show brisk mitotic rate (up to 10 mitotic figures per high power field); however, lacks pleomorphism
Differential Diagnosis22
- Peripheral giant cell granuloma
- Peripheral ossifying fibroma
- Fibroma
- Peripheral odontogenic fibroma
- Haemangioma
- Kaposi’s sarcoma
Diagnosis
- Diagnosis made on histopathological evaluation
Treatment
- Surgical excision of gingival lesions
- Curettage of underlying tissue
- Remove causative agent
- Recurrence rate of 15.8%
Neural Tumours
Neurofibroma
Neurofibroma is a benign peripheral nerve sheath tumour composed of a variable mixture of Schwann, perineural-like, and fibroblastic cells.
-
Benign peripheral nerve sheath tumour. It may also contain cells with features intermediate between these types, all contained within a collagenous or myxoid matrix.
-
Can occur as a solitary lesion or as part of a generalized syndrome of neurofibromatosis.
-
Approximately 4-7% of cases display oral manifestations.
Pathogenesis23
- The pathogenesis of solitary neurofibromas not associated with Neurofibromatosis-1 (NF-1) is poorly understood.
- Somatic inactivation of the NF-1 gene (located on chromosome 17) leads to increased and abnormal production of neurofibromin.
- This inactivation leads to the activation of p21(ras) and p13, causing cellular proliferation of Schwann cells associated with neurofibroma.
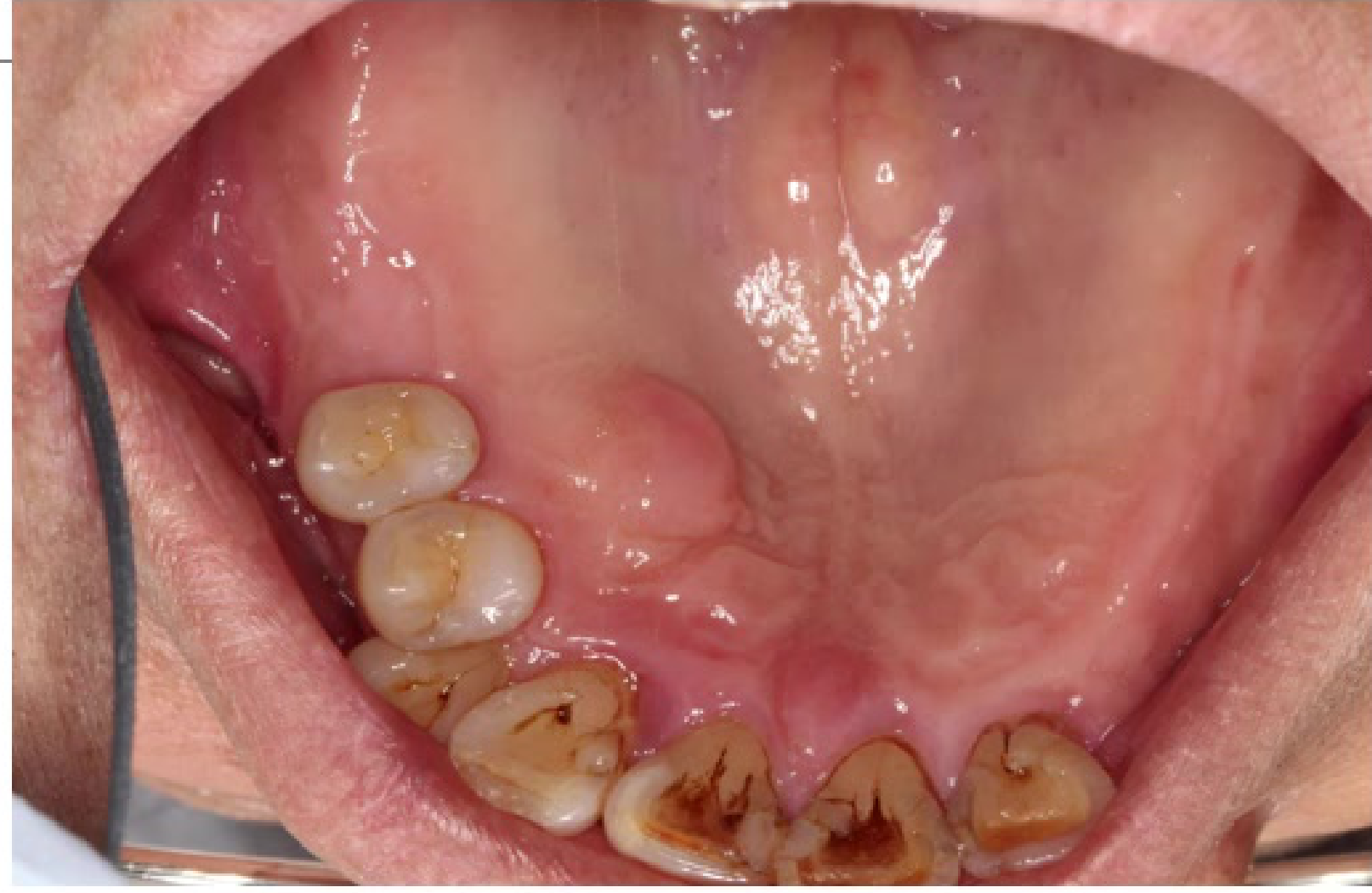
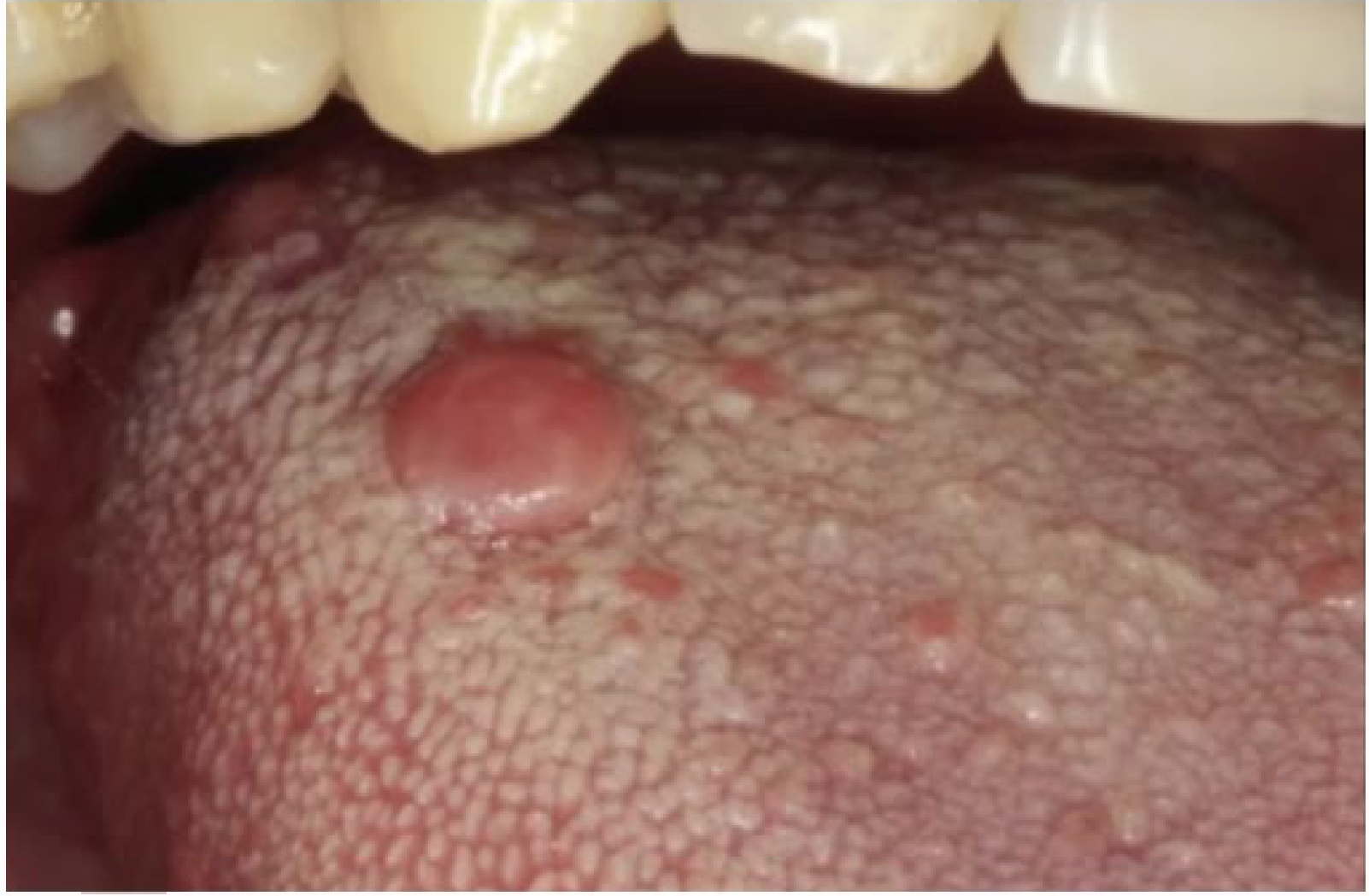
Clinical Features24
- In the oral cavity, these present as submucosal, nontender, discrete masses.
- Intraoral lesions of neural tissues mainly originate from branches of the fifth, seventh, and rarely the ninth cranial nerves.
- Common sites include the tongue, buccal mucosa, and vestibular area. The posterior mandible is the most common intraosseous location.
- Further divided based on clinical presentation - localised/solitary growth, diffuse discrete, multiple nodules, plexiform types
- overlying mucosa of solitary NF not associated with NF-1 gradually blends with surrounding normal mucosa, no clear-cut demarcation between lesion and normal mucosa
 |  |
Classification by Presentation
Neurofibromas are further divided based on clinical presentation:
- Localised/solitary growth
- Diffuse discrete
- Multiple nodules
- Plexiform types
In solitary neurofibromas not associated with NF-1, the overlying mucosa gradually blends with surrounding normal mucosa, with no clear-cut demarcation between the lesion and normal tissue.
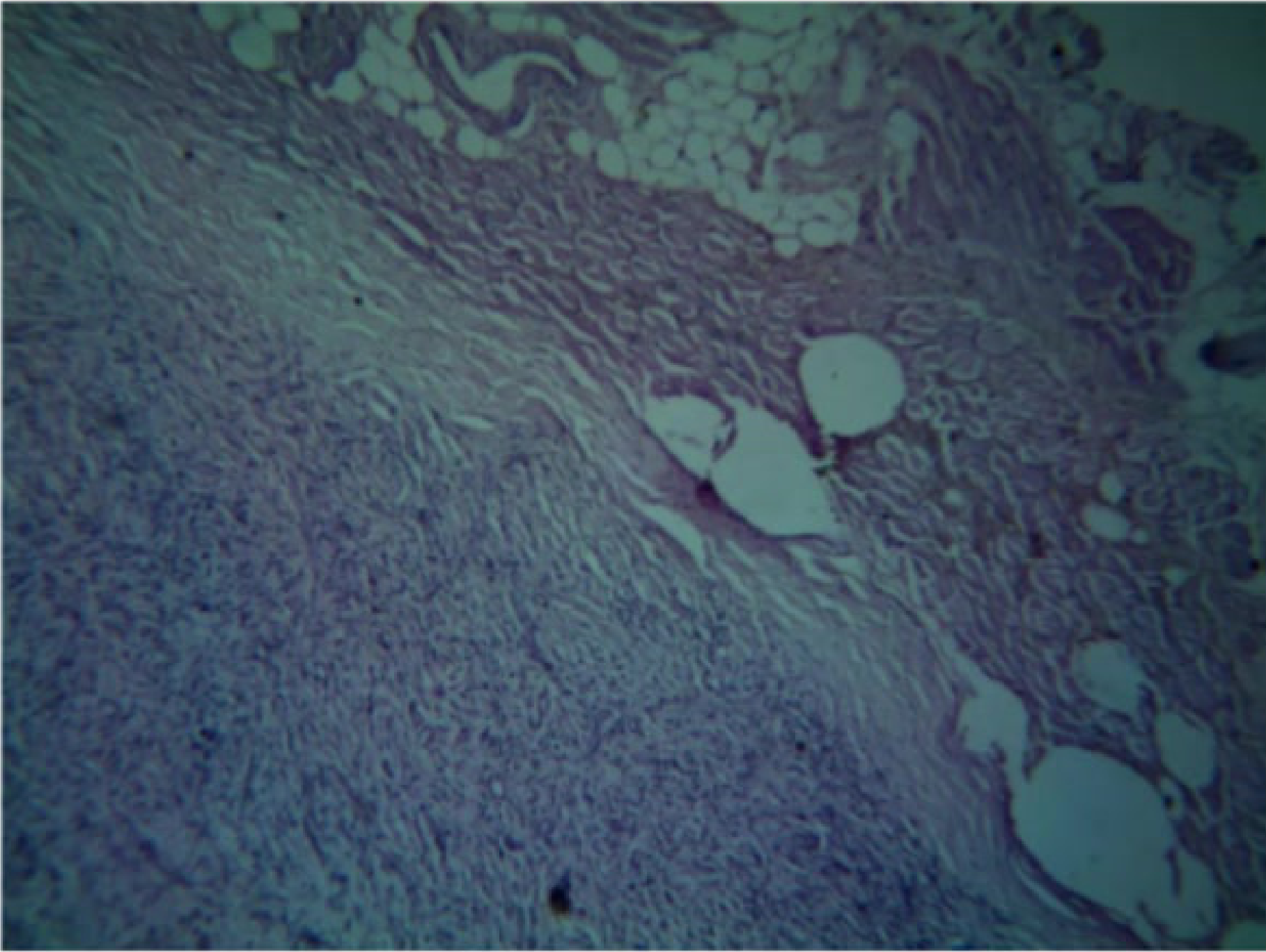
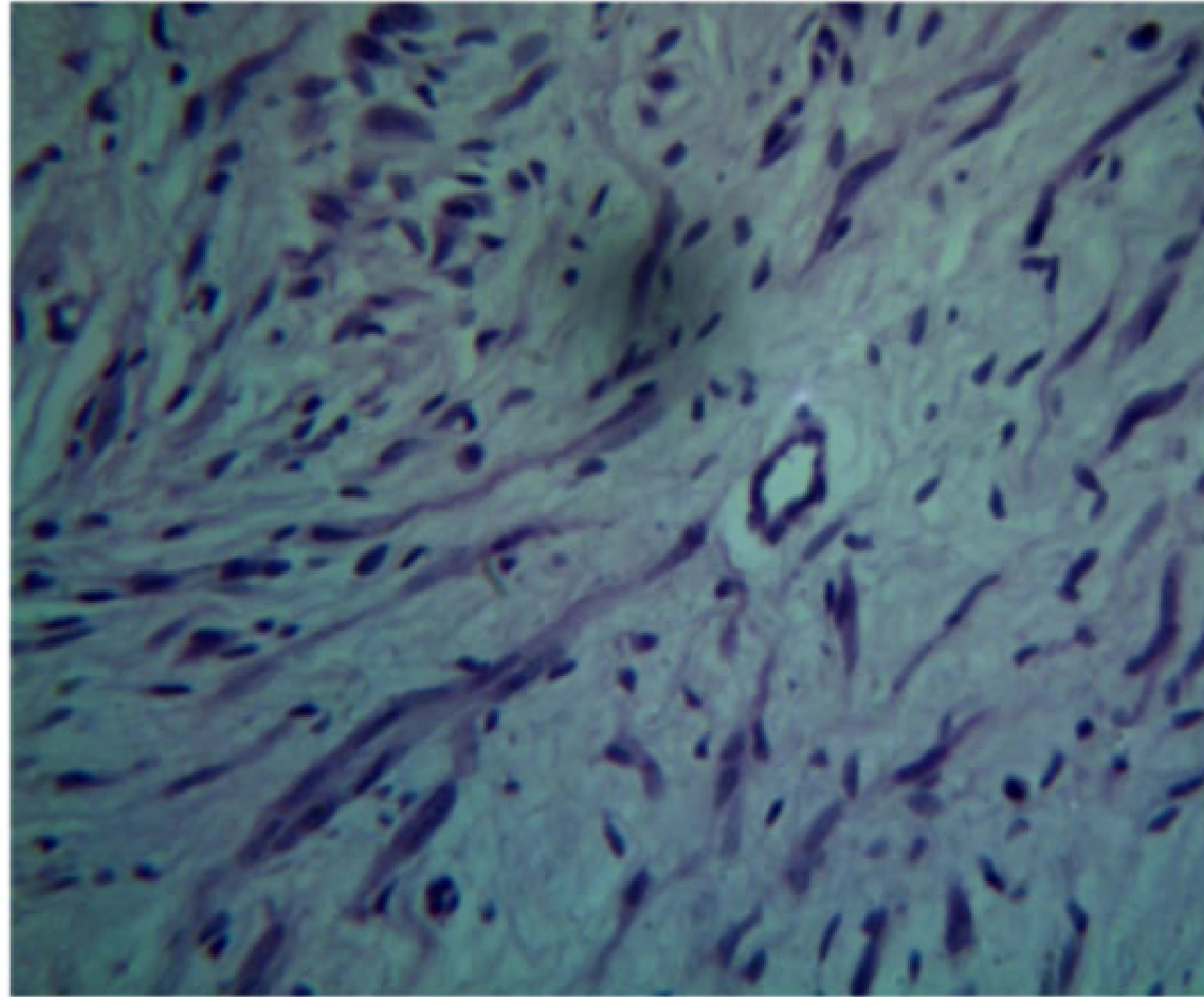
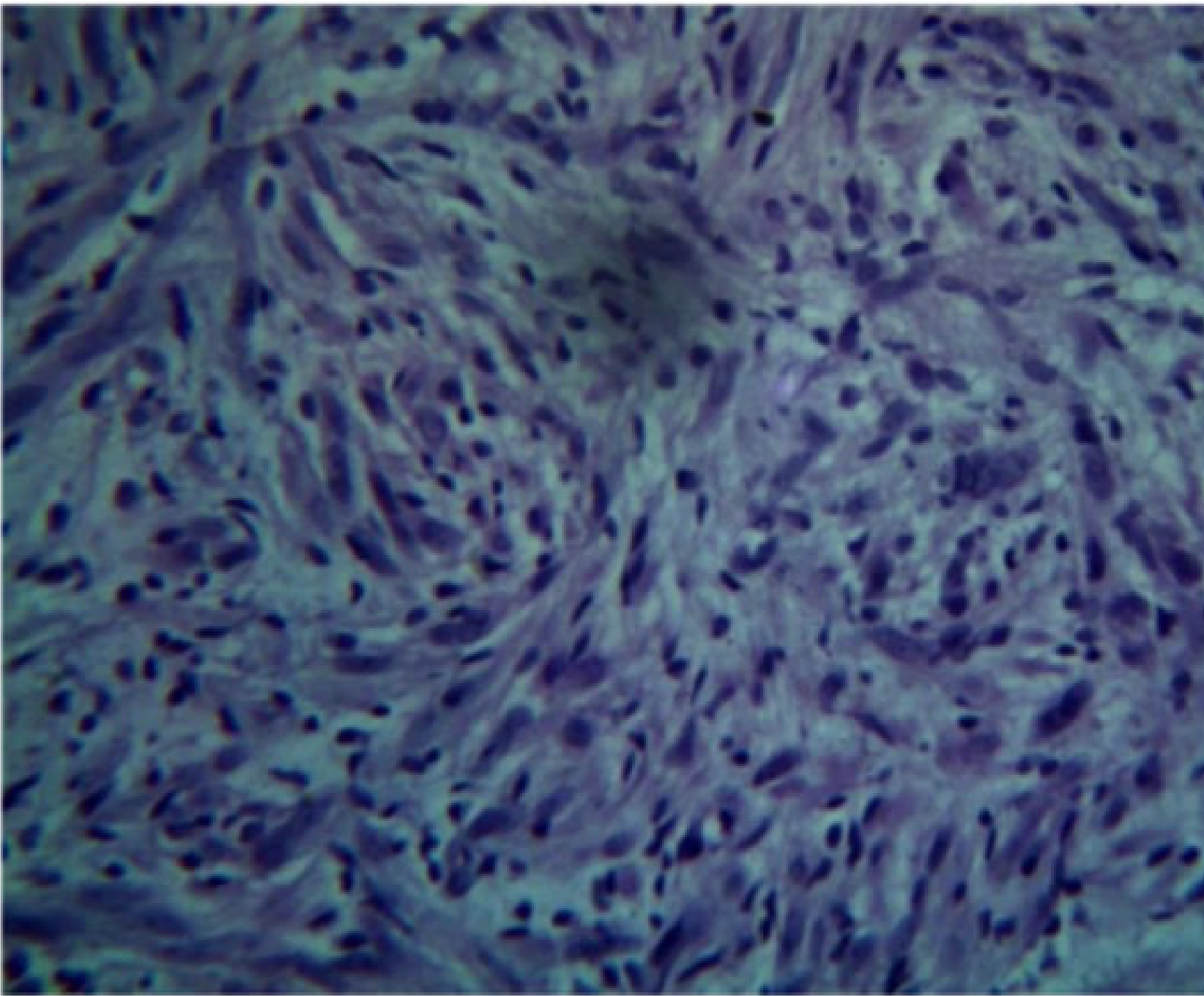
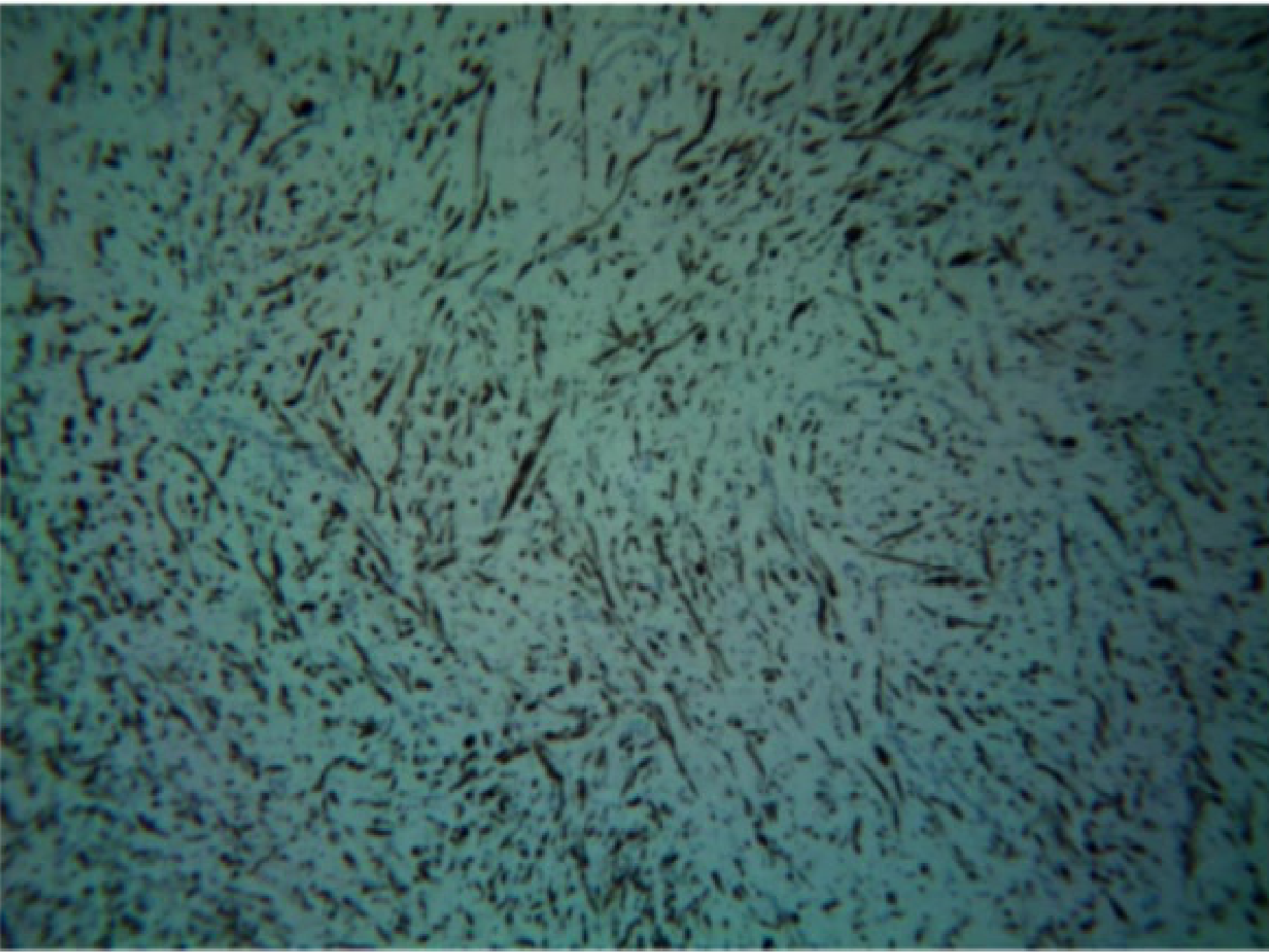
Histopathology25
- Unencapsulated lesion.
- Proliferation of Schwann cells.
- Elongated fibroblasts with wavy nuclei separated by abundant collagen fibres.
- Wavy nuclei separated by collagen fibers in a myxoid matrix.
- Presence of perineurial cells and mast cells.
- Myxoid connective tissue component.
 |  |
 |  |
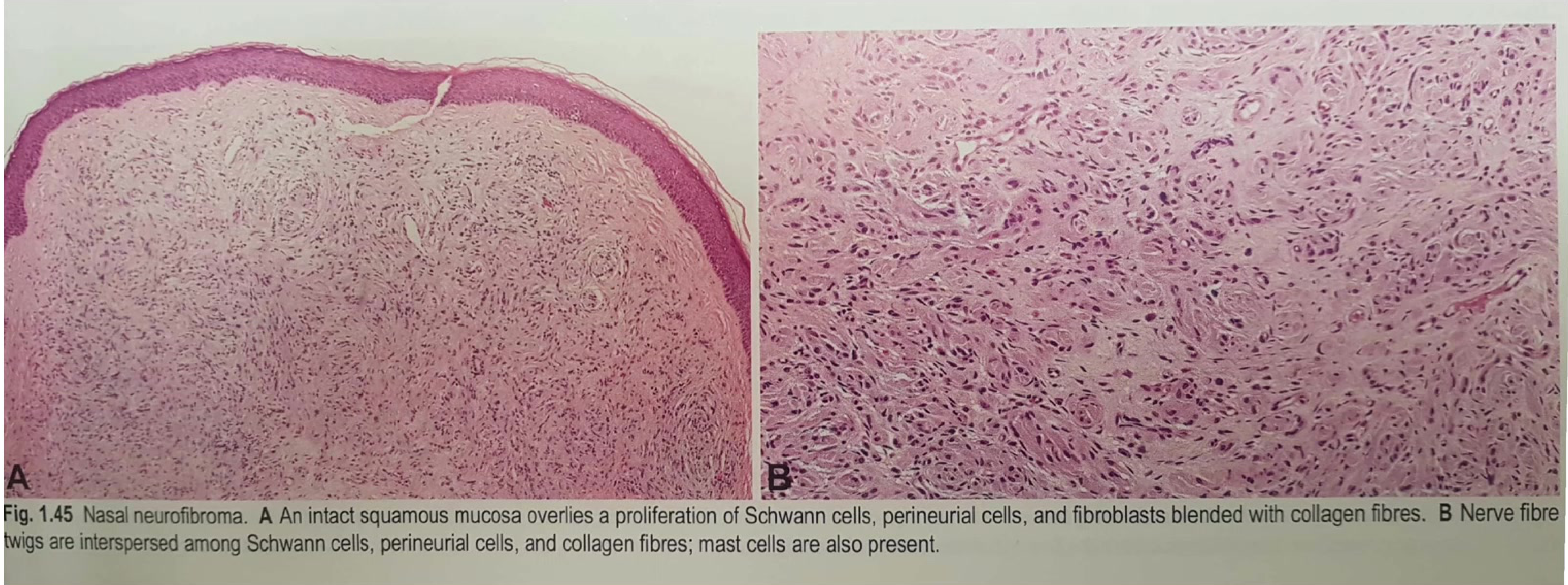
Microscopic Observations26
- Nasal Neurofibroma: Features an intact squamous mucosa overlying a proliferation of Schwann cells, perineurial cells, and fibroblasts blended with collagen fibres.
- Cellular Composition: Nerve fibre twigs are often interspersed among the Schwann cells and perineurial cells; mast cells are typically present within the matrix.
Neurofibromatosis Type 1
Neurofibromatosis type 1 (NF-1), previously known as von Recklinghausen disease, is a complex autosomal dominant disorder affecting multiple organ systems. It is caused by germline mutations in the NF1 tumour suppressor gene on chromosome 17. The defining feature of NF-1 is the neurofibroma.
Clinical Manifestations27
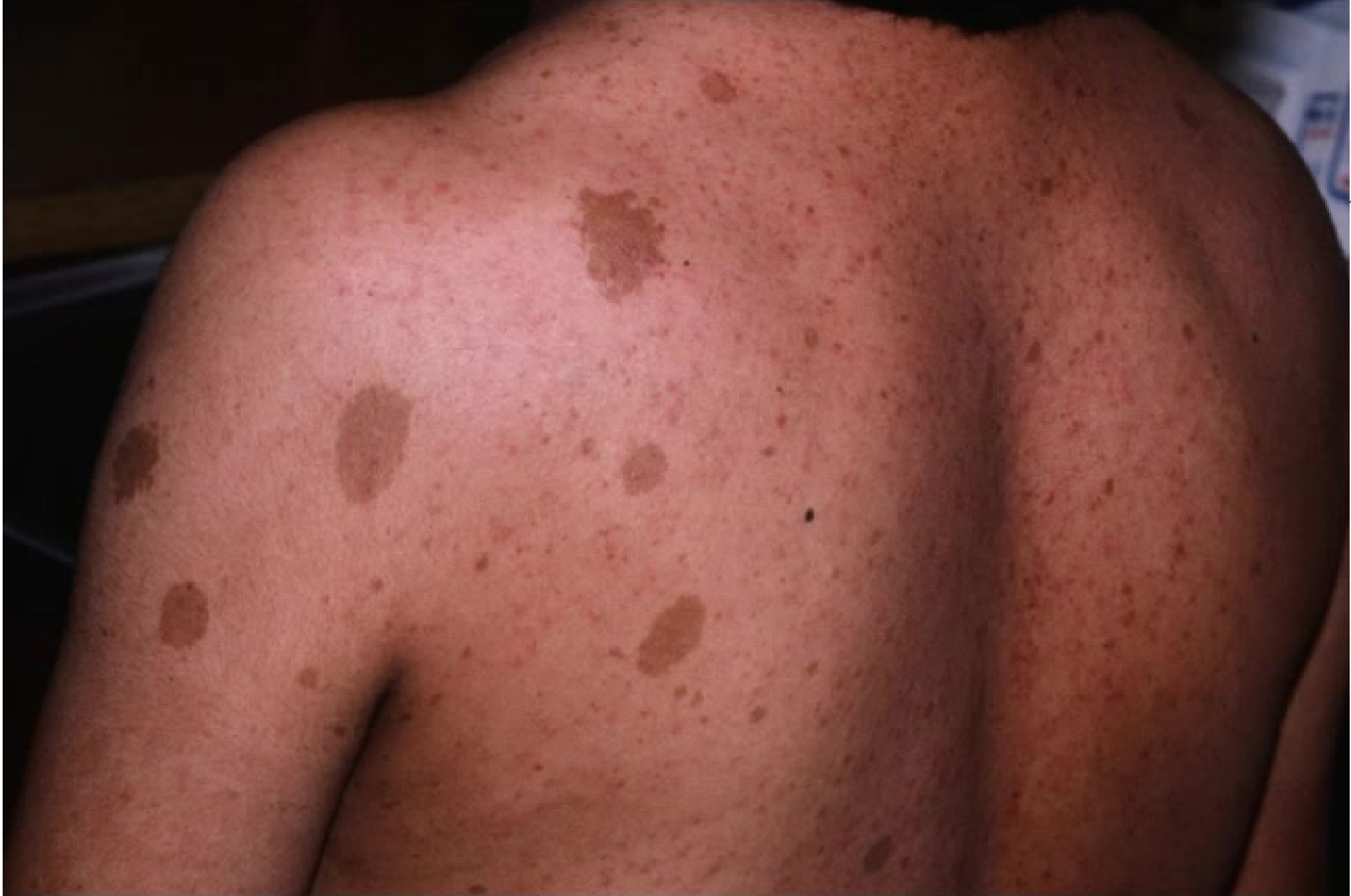
Nearly all individuals with NF-1 develop pigmented lesions and other systemic features: Pigmented features:
- Café-au-lait macules
- Skinfold freckling
- Lisch nodules
- Dermal neurofibromas
systemic features:
- Axillary freckling (Crowe’s sign).
- Skeletal abnormalities
- Brain tumours and peripheral nerve sheath tumours
- Learning disabilities, social, and behavioural problems resulting in lowered quality of life (QOL)
The disease is progressive over a lifetime, though the rate of progression and severity vary between individuals.
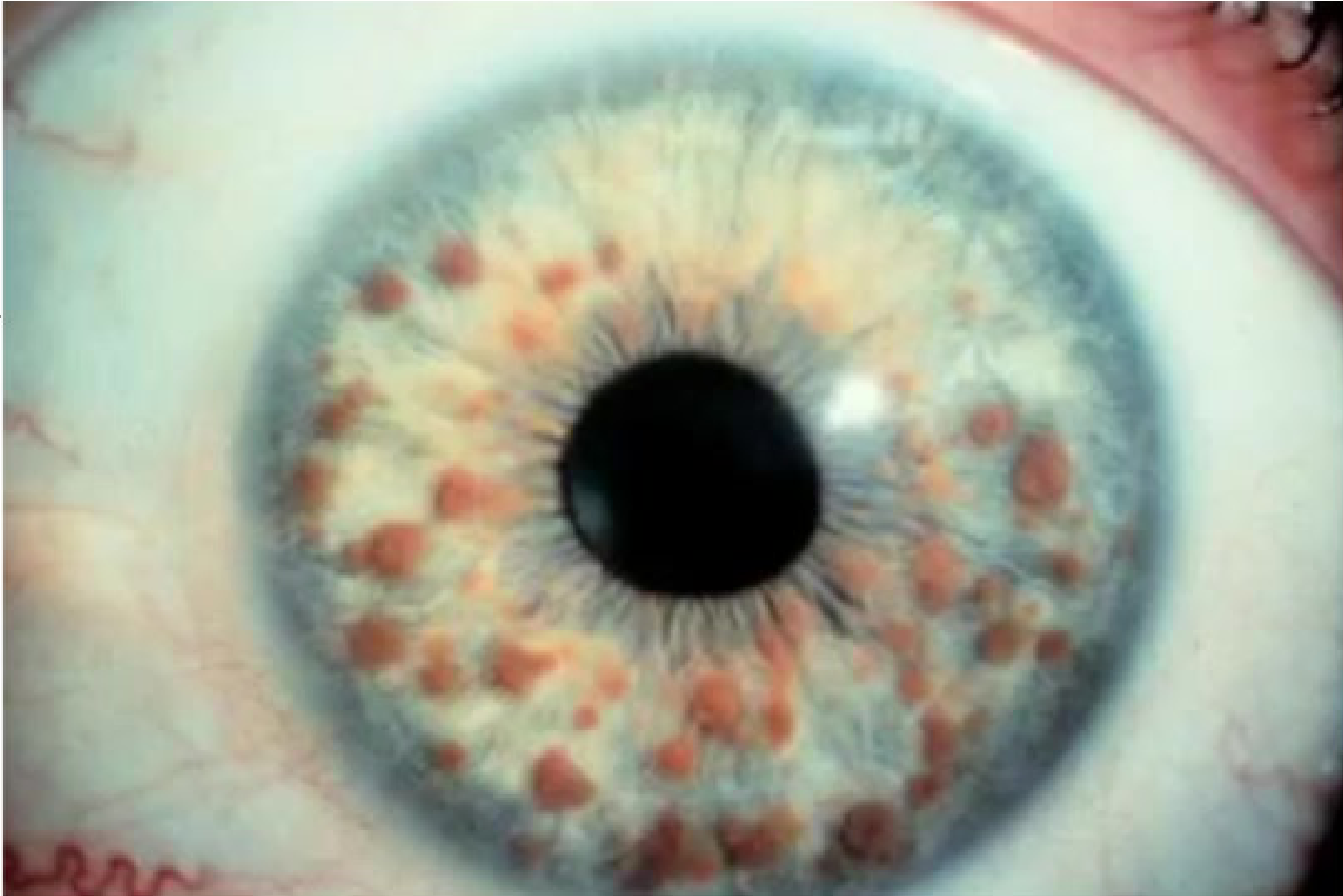
Ocular Findings28
- Lisch Nodules: A typical clinical appearance in patients with neurofibromatosis-1 involves the presence of multiple Lisch nodules in the iris
Lisch Nodules
These are translucent brown hamartomas found on the iris. .
 |  |
 |
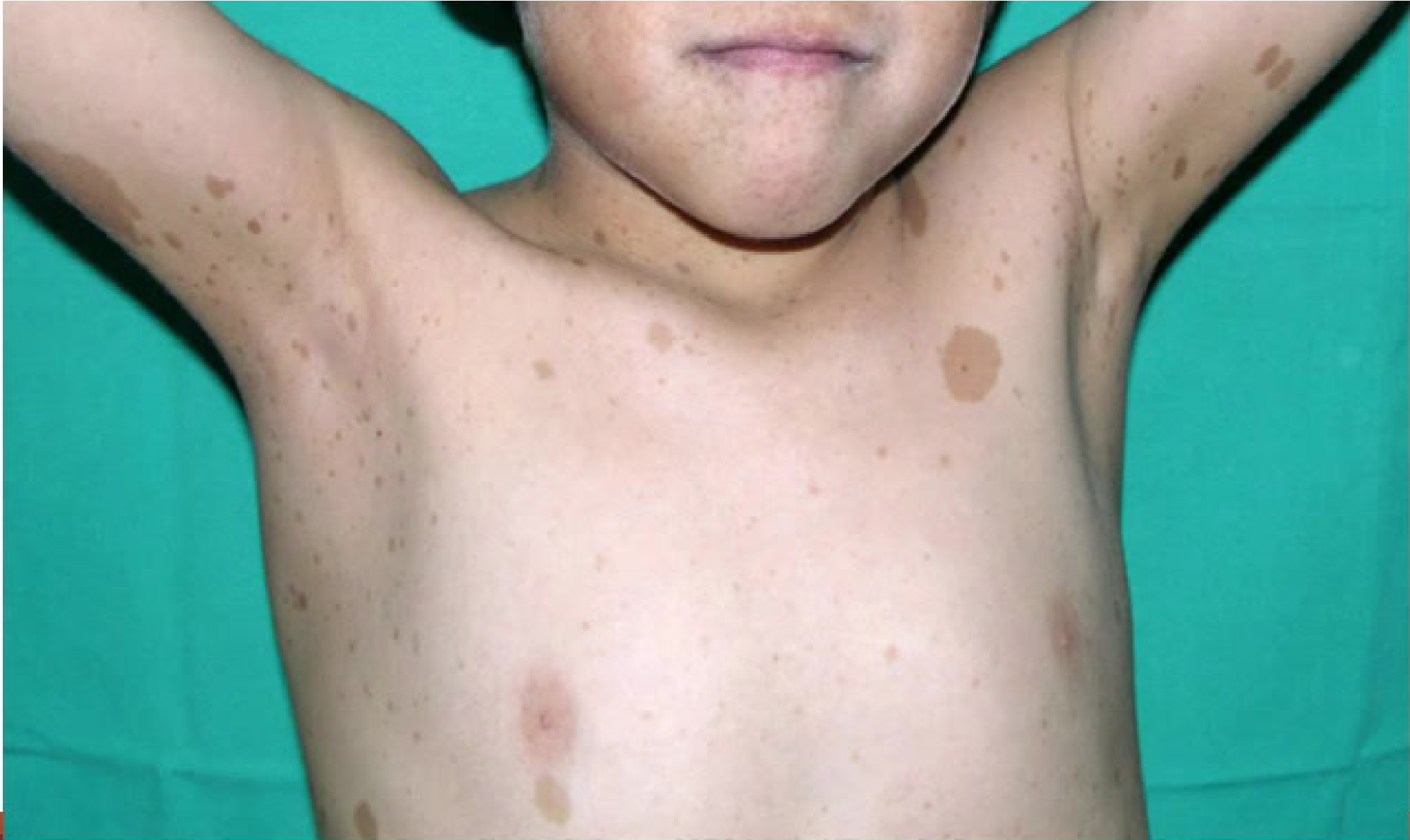
Systemic Involvement29
Manifestations of NF-1 include:
- Multiple skin nodules (dermal neurofibromas).
- Café-au-lait macules, often observed on the limbs.
- Internal involvement, such as spinal lesions detectable via MRI.
Epidemiology and Prognosis30
- Global Prevalence: 1 case per 3,000 individuals.
- Inheritance: 50% of cases are familial; the remainder arise from de novo NF1 mutations.
- Life Expectancy: Reduced by 8–21 years.
Malignancy Risks
- The most common cause of death in younger individuals is malignant peripheral nerve sheath tumour (MPNST).
- Cumulative risk of malignancy by age 50 is approximately 20–39%.
- Lifetime cancer risk is approximately 60%.
- There is a 50-fold increased risk for high-grade tumours, including malignant brain tumours and endocrine cancers.
- There is a 1000-fold increased risk for MPNSTs.
- Increased risks for buccal cavity, pharyngeal, oesophageal, skin (melanoma), thyroid, and ovarian cancers have also been described.
Approximately 72% of affected individuals exhibit oral manifestations.
Oral Manifestations of NF1
Gingival Enlargement and Pigmentation31
- Common in children with NF1.
- Presents as diffuse, unilateral enlargement of the attached gingiva.
- The tissue is fibrous and does not exhibit signs of inflammation.
- Rare cases may show melanin pigmentation of the gingiva.
Dental Abnormalities
- Impacted, supernumerary, missing, or displaced teeth.
- Plexiform neurofibromas can be associated with aplasia of mandibular second molars, increased spacing between teeth, and jaw asymmetries.
Soft Tissue and Osseous Lesions
- Neurofibromas: Commonly affect the tongue; plexiform neurofibromas may result in macroglossia.
- Osseous Lesions: Not very common but unique to the individual. Features may include increased size of the coronoid notch and lateral bowing of the ramus.
- TMJ: Neurofibroma involving the articular disc of the temporomandibular joint has been reported.
Diagnostic Criteria32
Solitary Lesions:
- Diagnosed via histopathological evaluation.
NF-1 Clinical Diagnosis: Based on the 1987 National Institutes of Health Consensus Conference, diagnosis requires at least two of the following major features:
- A first-degree relative with NF1.
- Six or more café-au-lait patches (>5 mm in prepubertal individuals; >15 mm in postpubertal individuals).
- Axillary or groin freckling.
- Two or more neurofibromas or one plexiform neurofibroma.
- Two or more Lisch nodules in the iris.
- Optic pathway glioma.
- A distinctive osseous lesion (e.g., bony dysplasia of the sphenoid wing or pseudoarthrosis of the long bones).
Management of Solitary Neurofibroma33
- Complete excision is the standard treatment.
- There is a 5% recurrence rate, usually due to incomplete excision.
- Malignant transformation is rare.
Management of NF-1 Complications
Early detection of treatable complications is essential.
- Dermal Neurofibromas: Surgical removal, laser ablation for small lesions, or electrodesiccation.
- Plexiform Neurofibromas: Pain management and excision of surgically amenable tumours. Biologically targeted therapies (mTOR inhibitors, imatinib, selective MEK inhibitors) are being evaluated.
- Atypical Neurofibromas: Symptomatic lesions showing hypercellularity and atypical nuclei are suggestive of pre-malignancy. These require complete excision and clinical surveillance.
- MPNST: Treated with complete excision and neoadjuvant chemotherapy.
Neurilemoma
Neurilemoma (Schwannoma) is the most common benign neurogenic neoplasm. It is a neoplastic proliferation comprised exclusively of cells that resemble Schwann cells and possess the corresponding antigenic phenotype.
Aetiology34
- 90% of cases are sporadic.
- Can occur in specific syndromes: NF2 (3%), Schwannomatosis (2%), and Carney complex.
- Genetic factors play a role.
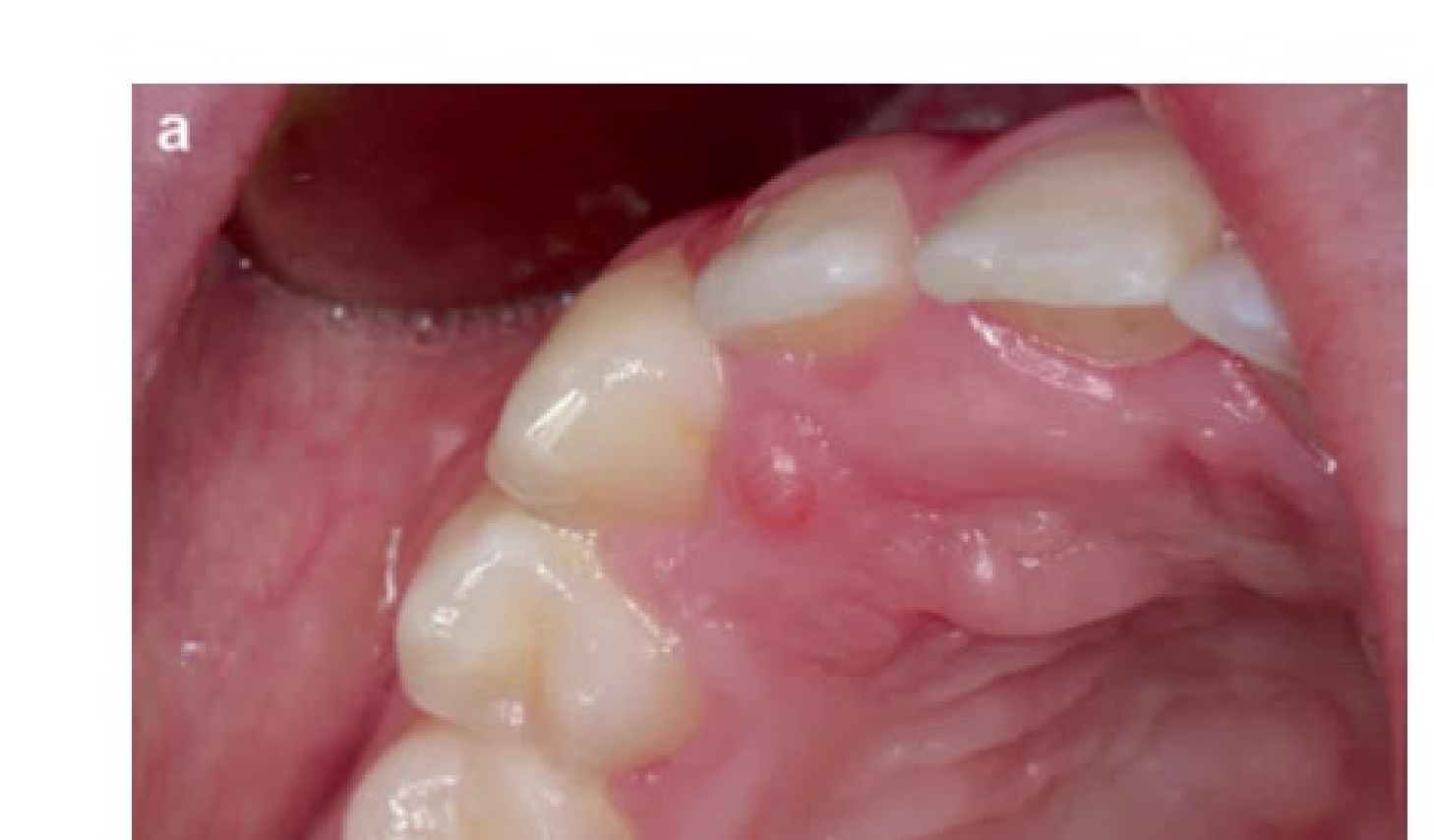
Clinical Presentation
- Majority are non-vestibular or extracranial.
- Typically presents as a solitary, encapsulated mass of long duration.
- Rapid growth is rare.
- Usually painless and not ulcerated.
- May manifest with facial hypoesthesia, paraesthesia, or pain.
- Bilateral schwannomas are specifically related to NF-2.
Neurofibromatosis Type 2
Neurofibromatosis type 2 (NF2) is a dominantly inherited tumour predisposition syndrome caused by mutations in the NF2 gene on chromosome 22. Although classified as a “neurofibromatosis,” actual neurofibromas are relatively infrequent in this condition.
Characteristic Tumours35
- Schwannomas
- Meningiomas
- Ependymomas
Clinical Features
- The majority of patients develop bilateral schwannomas involving the superior vestibular branch of the 8th cranial nerve.
Important Distinction
Despite the name, neurofibromas are not typically seen in NF2; the condition is characterized by schwannomas, meningiomas, and ependymomas.
- Symptoms include hearing loss, tinnitus, and imbalance.
Histopathology36
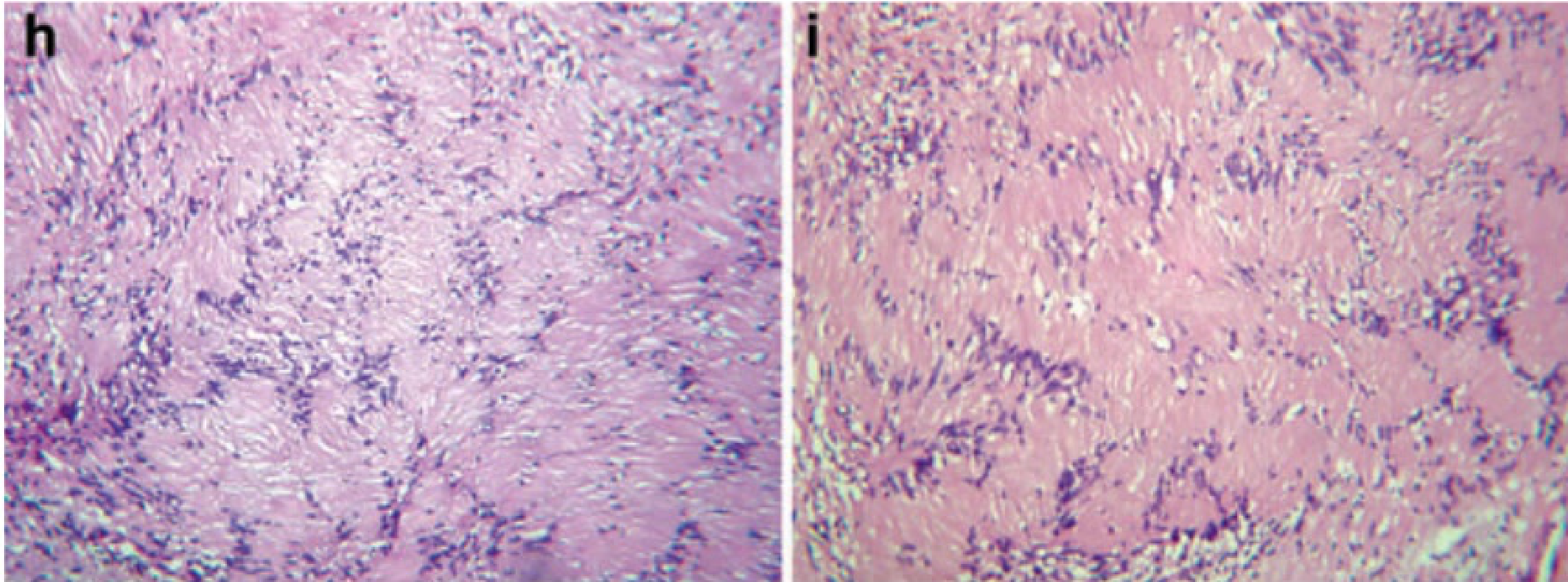
Neurilemomas are encapsulated and typically exhibit two main patterns:
- Antoni A: Highly cellular area composed of elongated Schwann cells. It features organized spindle-shaped cells in a palisading arrangement around acellular, eosinophilic areas known as Verocay bodies.
- Antoni B: Composed of elongated Schwann cells arranged in a less dense, more disorganized myxoid manner compared to Antoni A.
As the tumour enlarges, cystic change becomes more prominent, often associated with mucinous degeneration, haemorrhage, necrosis, and microcystic formation.
Diagnosis37
- Diagnosis is confirmed through histopathological evaluation.
Management
- Complete resection is curative; the lesion is not likely to recur.
- Malignant transformation has not been reported.
- Depending on the location and potential risk to major nerves or vital structures, clinicians may opt for close follow-up observation instead of immediate surgery.
Adipose Tumours
Lipoma
Lipoma is a rare, benign tumour of the adipose tissue.
Epidemiology and Prevalence38
- Most frequent benign mesenchymal tumour.
- More common in men.
- Often seen in overweight individuals.
- 15-20% of cases occur in the head and neck region.
- Intra-oral lesions are less frequent, representing 1-4% of head and neck lipomas.
- Relatively rare intraorally, representing only 1–4% of all oral tumours.
Aetiopathogenesis
- These lesions are well-circumscribed benign neoplastic proliferations of adipose tissue.
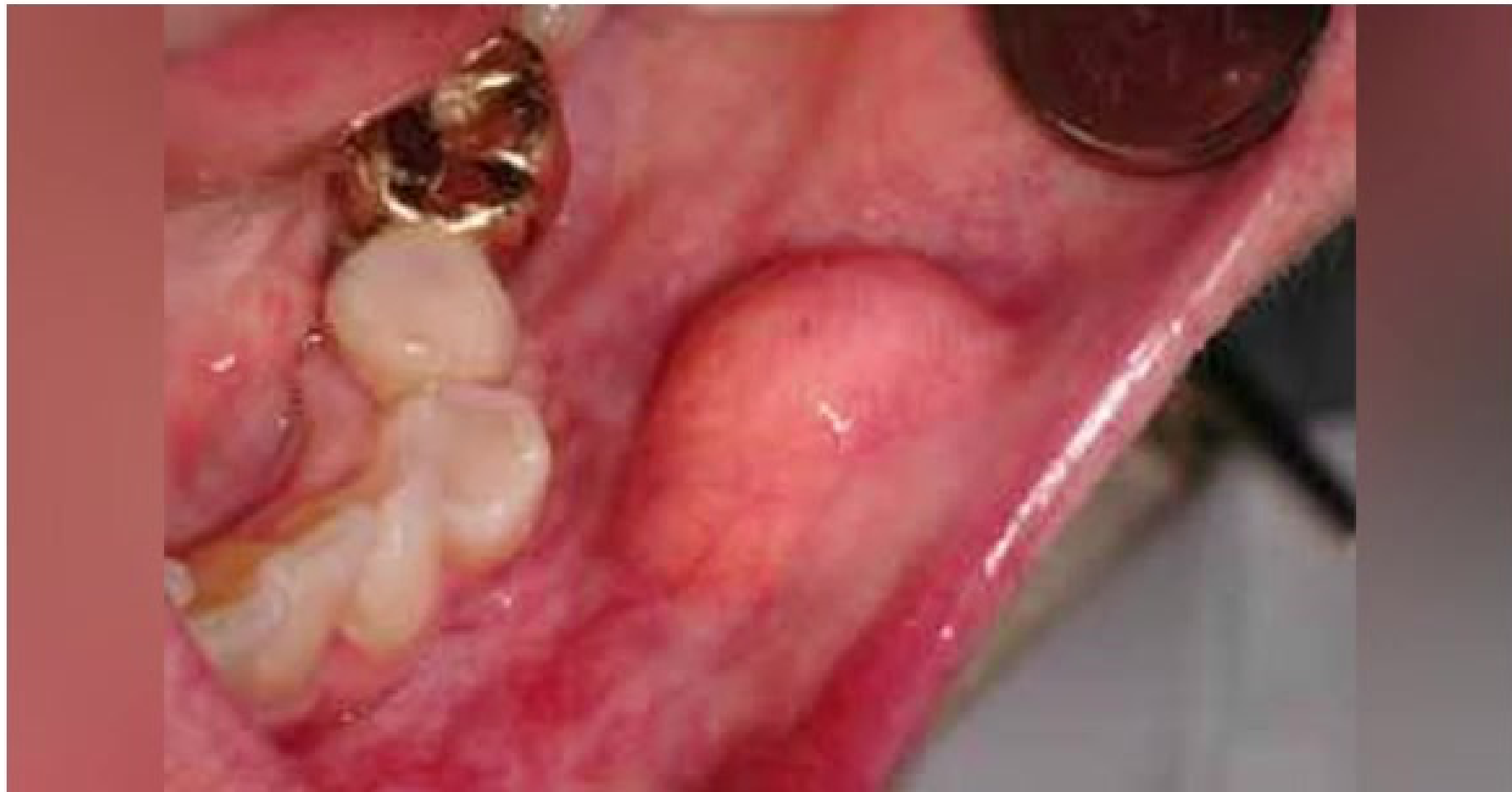
Clinical Presentation39
- Physical Characteristics: Slow-growing, yellowish, soft, semi-fluctuant, and painless mass.
- Common Sites: Usually seen on the buccal mucosa, followed by the tongue, lips, and floor of the mouth.
- Appearance: Covered by normal mucosa or skin; can be solitary or multiple.
- Size: Lesions vary in size, with an average diameter of 2cm.
 | 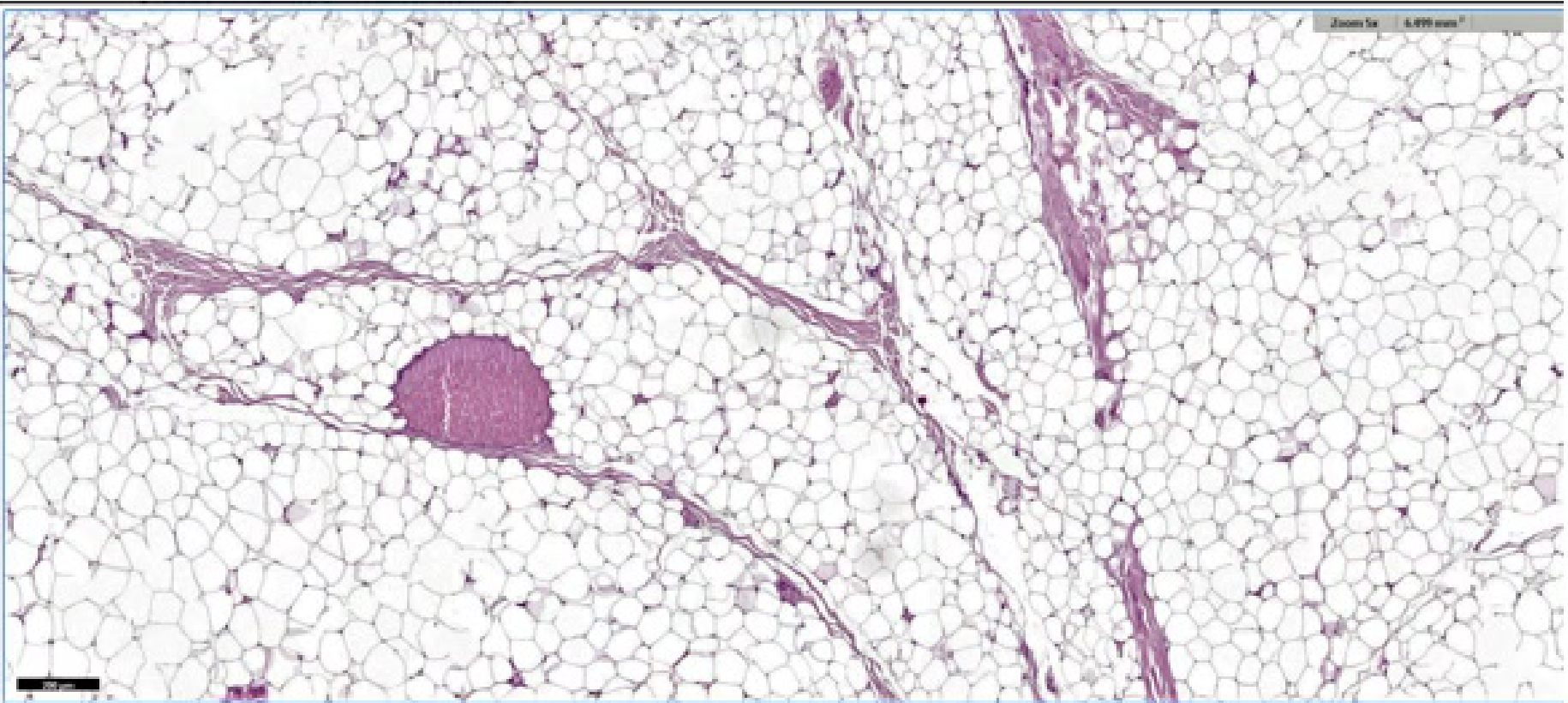 |
Histopathology
- Characterized by the proliferation of mature adipocytes.
- The proliferation is typically surrounded by a thin fibrous capsule.
- Paucicellular fibrous septa may be present.
- Fat necrosis is often found in larger tumours.
Diagnostic Evaluation
- Imaging: Lesions demonstrate characteristic low attenuation on CT. Signal intensity is similar to that of fat on all MRI sequences.
- Biopsy: A diagnostic biopsy typically exhibits a connective tissue capsule circumscribing lobules of hexagonal cells that resemble normal adipose tissue.
- Systemic Considerations: Patients presenting with multiple lesions throughout the body should be investigated for syndromes and rare obesity disorders.
Management and Prognosis
- Surgical Intervention: Treatment consists of an excisional biopsy.
- Pre-surgical Planning: Adequate imaging, diagnostic biopsy, and careful assessment are paramount before planning surgery for large or difficult-to-access lesions.
- Recurrence: Recurrence is unlikely following excision.
Vascular Tumours
Hemangioma
Hemangiomas are true neoplasms of blood vessels formed by endothelial proliferation assuming variable width.
Aetiology and Pathogenesis40
- Hemangiomas originate from embryonic placental angioblasts or intrinsic endothelial progenitor cells. These cells possess the ability to clonally duplicate within a precise environment influenced by cytokines and oestrogen concentrations.
- They arise from progenitor cells with a directional preponderance to become placental-like tissue in specific organs, such as the skin and liver.
- Molecular signalling plays a significant role in vascularization. During the proliferative phase of growth, increased levels of molecular contributors to endothelial cell migration and new vessel development are present, including:
- Vascular endothelial growth factor (VEGF)
- Basic fibroblast growth factor
- Insulin-like growth factor
- Matrix metalloprotease-9
Clinical Presentation and Progression41
- Hemangiomas typically proliferate during the first 9–12 months of life and subsequently involute over a variable course lasting many years.
- There is a noted female predominance.
- Infantile hemangiomas develop shortly after birth, with 60% occurring in the head and neck region. They appear as well-demarcated, red, vertically expansive lesions.
- Congenital hemangiomas are rare, present at birth, and do not follow the natural growth phase of infantile types.
- Physical characteristics include smooth, reddish, purple, sessile, polypoid, or pedunculated masses. These often increase in size and may exhibit occasional bleeding.
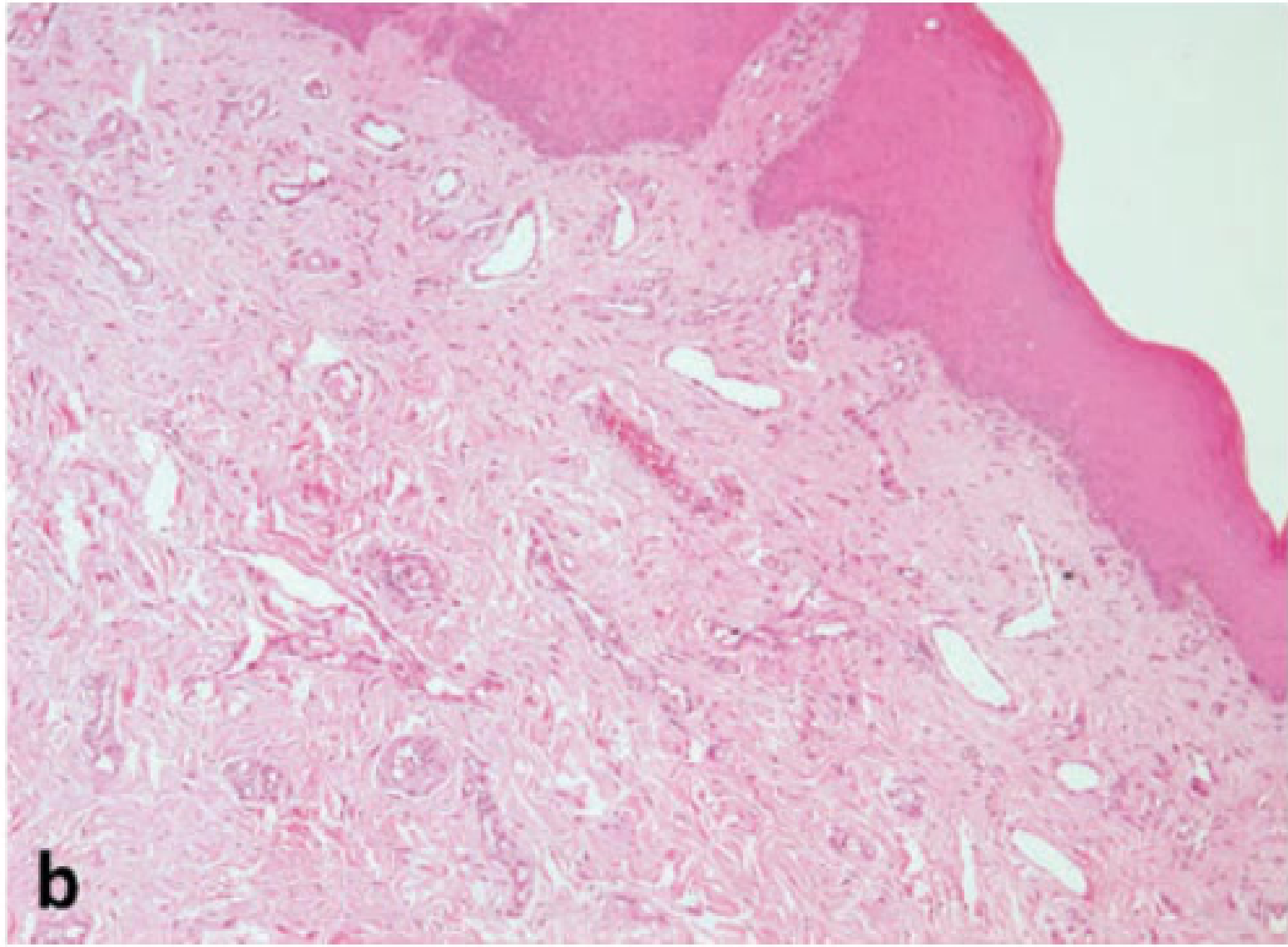
Histopathological Classification42
Hemangiomas are classified according to the predominant lumina of the capillary vessels within the lesion.
- Capillary Hemangiomas: Consist of multilobular arrangements of proliferating endothelial cells and capillaries of various shapes and sizes, surrounded by pericytes. These appear more cellularly rich, though mitotic activity is similar to the cavernous type.
- Cavernous Hemangiomas: Found in the penial cavernous body, these show larger dilated vascular spaces lined by endothelial cells.
Diagnostic Confirmation
- A final diagnosis may require an immunohistochemical panel to differentiate the vascular structure.
- Diagnosis is often confirmed by a history of rapid growth followed by a period of stabilization and eventual involution.
Diagnostic Approach43
- History and Clinical Examination: Includes the use of diascopy.
- Biopsy: Generally NOT indicated due to the significant risk of bleeding.
- Investigations: May include specialized imaging or angiography.
Management Strategies
- Observation: Small hemangiomas often require no treatment as they typically self-involute by age 9.
- Laser Therapy: Utilized for specific lesion types or locations.
- Pharmacotherapy: Use of corticosteroids.
Malignant Soft Tissue Tumours
Rhabdomyosarcoma
Rhabdomyosarcoma represents a group of malignant neoplasms derived from skeletal striated muscle cells. It is recognized as the most common soft tissue sarcoma in children and adolescents, accounting for approximately 5-8% of all childhood malignancies.
Aetiology and Genetic Associations44
While most cases present sporadically, a small subset of patients develops the condition as part of a genetic syndrome, including:
- Beckwith-Wiedemann syndrome
- Von Recklinghausen disease (Neurofibromatosis type 1)
- Gorlin syndrome (Basal cell nevus syndrome)
Clinical Features45
Clinical presentation typically involves a palpable, circumscribed, and rapidly growing mass. The disease is categorized into three subsites based on anatomic location and the risk of local relapse:
- Parameningeal sites: including the paranasal sinuses, nasopharynx, nasal cavity, and pterygopalatine fossa.
- Oral cavity sites.
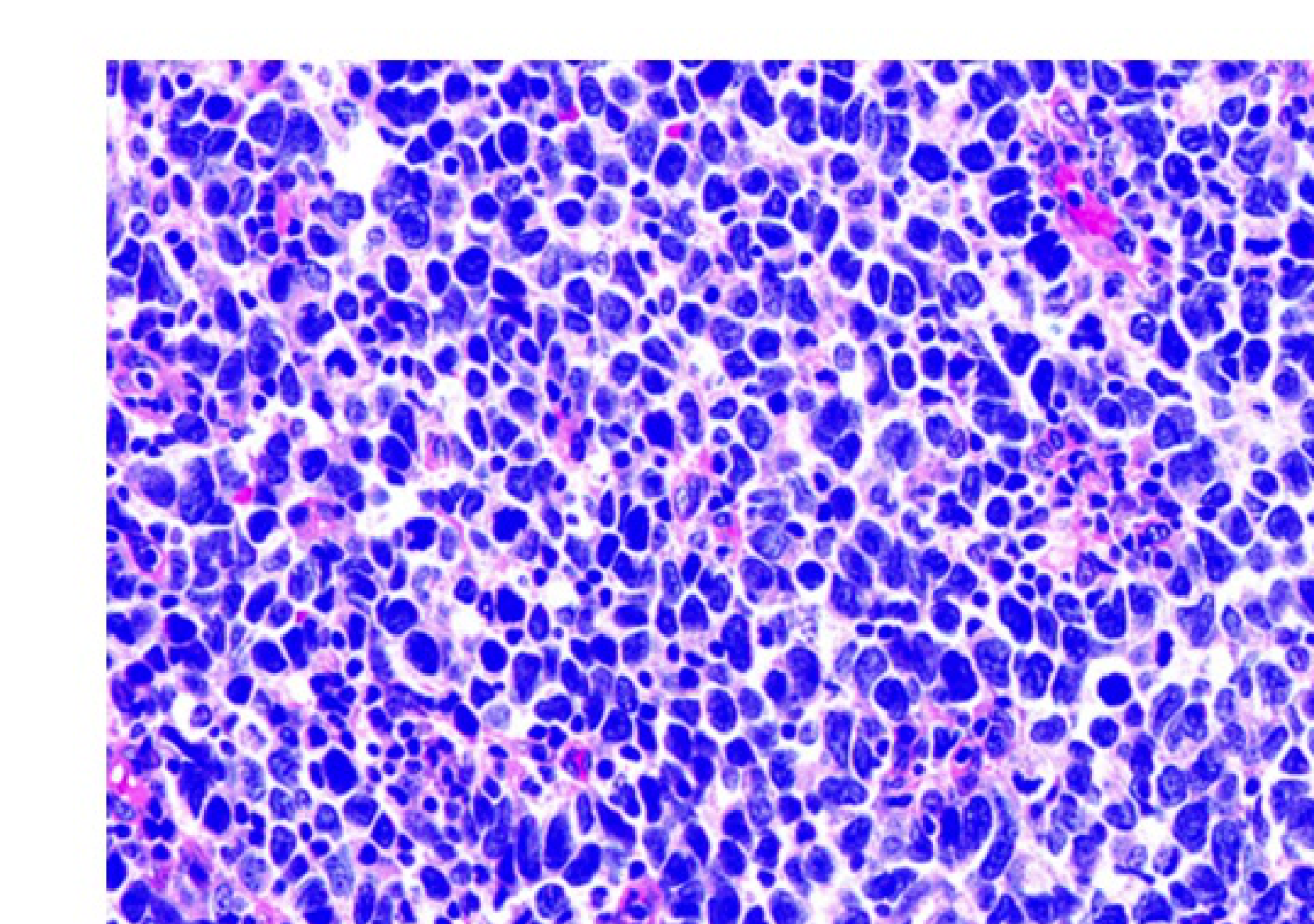
Histopathology
There are three primary microscopic subtypes of rhabdomyosarcoma:
- Alveolar: Characterized by sheets of small, round cells clustered with variable amounts of fibrous septa. Scattered giant cells may be present.
- Embryonal: Features cytologically round to spindle cells with scant cytoplasm set within a myxoid background. Elongated cells with more abundant eosinophilic cytoplasm are referred to as “strap” or “tadpole” cells.
- Pleomorphic: Consists of sheets of large cells demonstrating marked nuclear atypia or “anaplasia” with eosinophilic cytoplasm.
Diagnosis46
Diagnosis is established through comprehensive histopathological assessment.
Treatment and Prognosis
Management of rhabdomyosarcoma is challenging due to an increased risk of local control failure. The condition is characterized by a high rate of early metastases and recurrence. Treatment typically involves a multimodal approach:
- Surgery
- Chemotherapy
- Radiotherapy
Fibrosarcoma
Fibrosarcoma is a malignant neoplasm of mesenchymal origin where fibroblasts serve as the cell of origin. These tumors can arise within soft tissue or within the bone.
Clinical Presentation47
Common clinical signs and symptoms include:
- Pain and swelling
- Paraesthesia
- Occasional loss of teeth
- Ulceration of the overlying mucosa
In some instances, the lesion may appear as an innocuous, lobulated, sessile, and painless submucosal mass of normal coloration without hemorrhage.
Progression and Metastasis
Fibrosarcoma is associated with a high rate of local recurrence. Hematogenous metastasis may occur, frequently involving the following sites:
- Lungs
- Mediastinum
- Abdominal cavity
- Bone
Microscopic Characteristics48
Fibrosarcoma is characterized by a highly cellular fibroblastic proliferation typically arranged in a “herringbone” pattern. This pattern consists of cells in columns of short parallel lines, where lines in one column slope in one direction while lines in adjacent columns slope the opposite way.
Key histological features include:
- Cells with scant cytoplasm and tapering, elongated dark nuclei.
- Increased granular chromatin and variable nucleoli.
- Presence of mitotic activity, often including abnormal forms.
- Variable amounts of collagen.
- Absence of giant cells and significant pleomorphism (the presence of which might suggest pleomorphic Malignant Fibrous Histiocytoma).
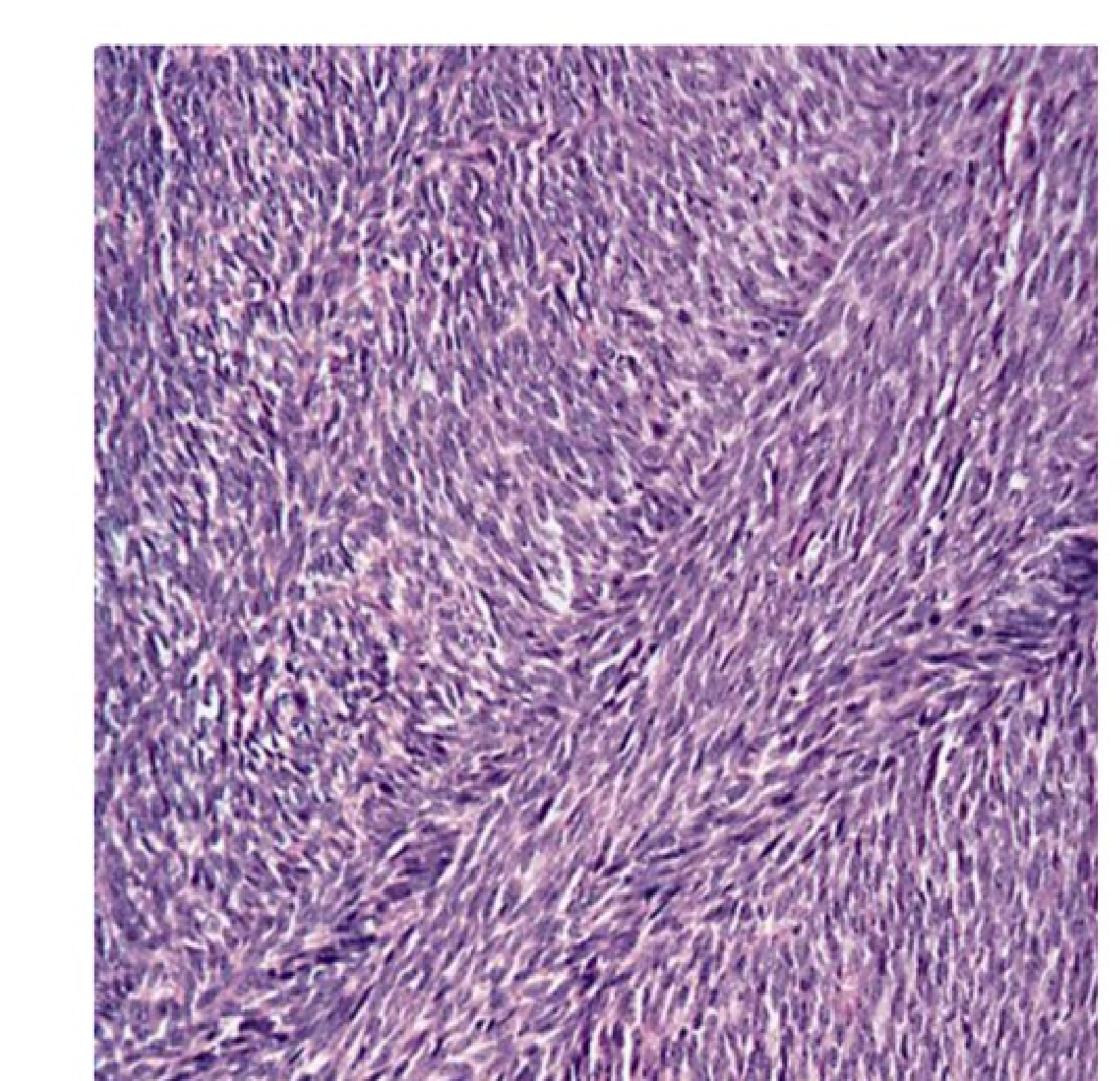
Histological Patterns
Observed growth patterns include:
- Keloid-like (featuring thick hyalinized collagen fibers)
- Loose fascicular
- Focally myxoid
Diagnostic Evaluation49
It is essential to determine the local extent of the neoplasm and the presence or absence of local and distant metastasis.
- Imaging: CT or MRI is utilized for staging.
- Grading: Histopathology classifies the tumor into low-grade and high-grade variants.
Management
Treatment strategies include:
- Wide local excision: Requiring at least a 1 cm margin confirmed by histopathologic clearance.
- Radiotherapy
- Chemotherapy
Kaposi Sarcoma
Kaposi Sarcoma is an angioproliferative malignant neoplasia of endothelial cells. It forms an infiltrative, capillary-rich lesion that eventually disseminates to multiple cutaneous sites, viscera, and lymph nodes.
Aetiology and Pathogenesis50
The condition is caused by Human Herpesvirus 8 (HHV8) infection of endothelial cells. While it can be found in immunocompetent individuals, it is significantly more active during immunosuppression.
In AIDS-related Kaposi Sarcoma (AIDS-KS), the Tat protein produced by HIV-infected lymphoid cells promotes HHV8 infection. This contributes to the highly aggressive nature of the disease by inducing inflammatory cytokines and angiogenesis.
Clinical Presentation and Epidemiology51
Kaposi Sarcoma is the second most frequent tumor in HIV-infected patients worldwide and the most common cancer in Sub-Saharan Africa.
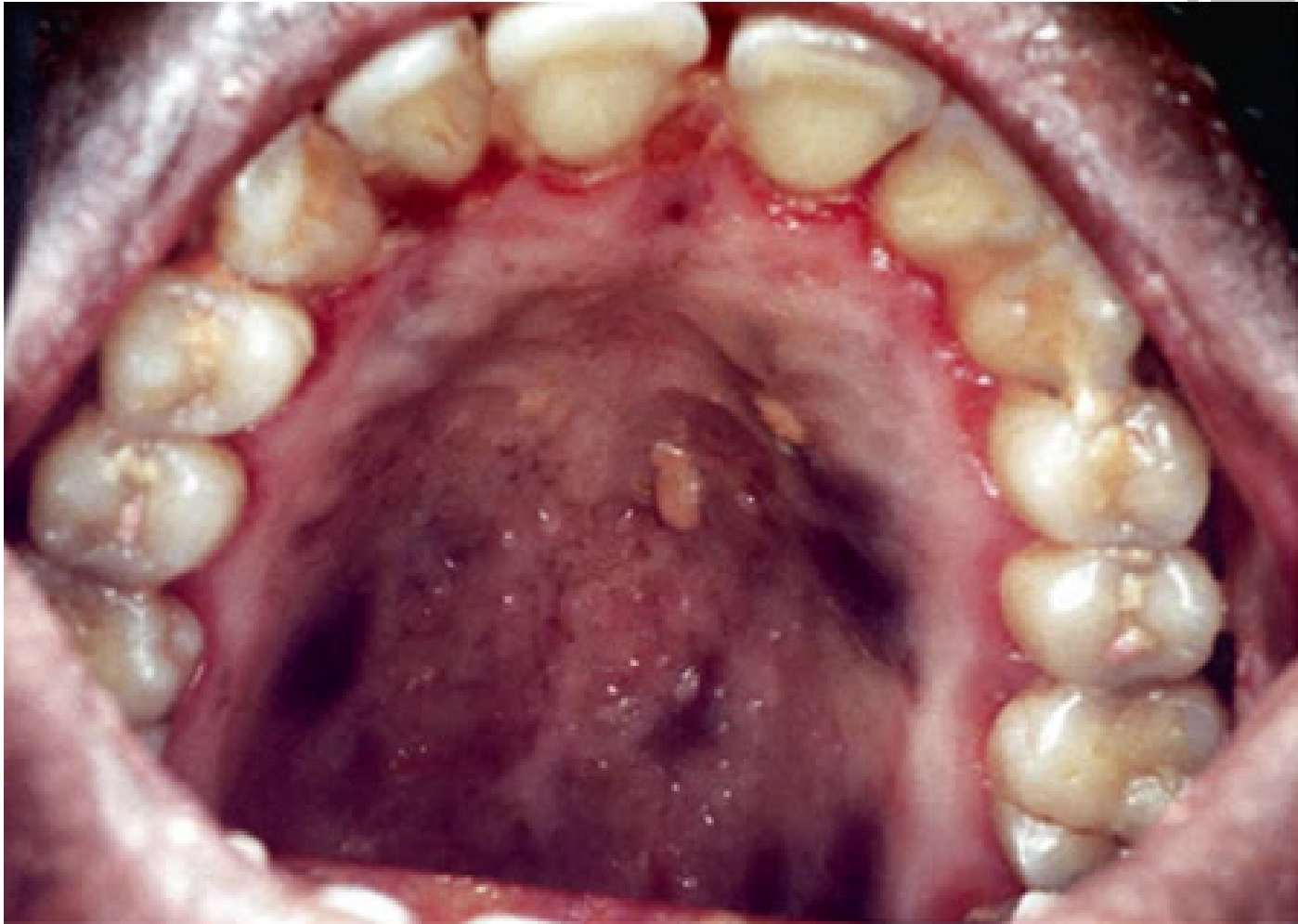
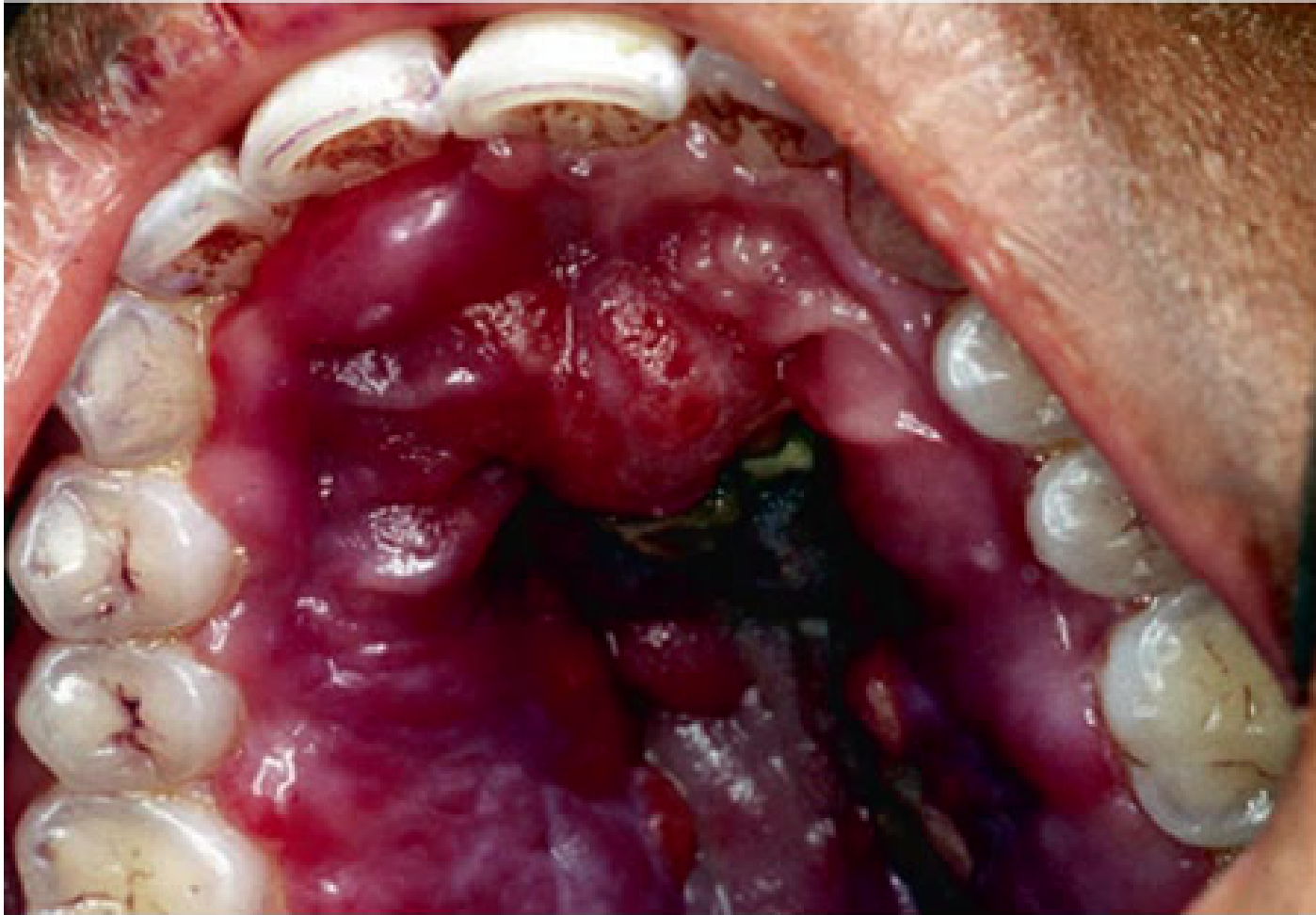
- Oral Manifestations: The most common oral locations are the hard palate, followed by the gingiva and the tongue. Approximately 70% of patients with cutaneous AIDS-related KS also present with oral lesions.
- Appearance: Common presentations include multiple patches and papules with a bluish to purple color. As the disease progresses, these lesions transition into a nodular stage.
- Advanced Symptoms: Advanced lesions may exhibit hemorrhage, pain, ulceration, and secondary infections.
- Aggressive Behavior: Highly aggressive lesions are infiltrative, involving both soft tissue and bone, and can disseminate to compromise visceral organs and lymphatic nodes.
 |  |
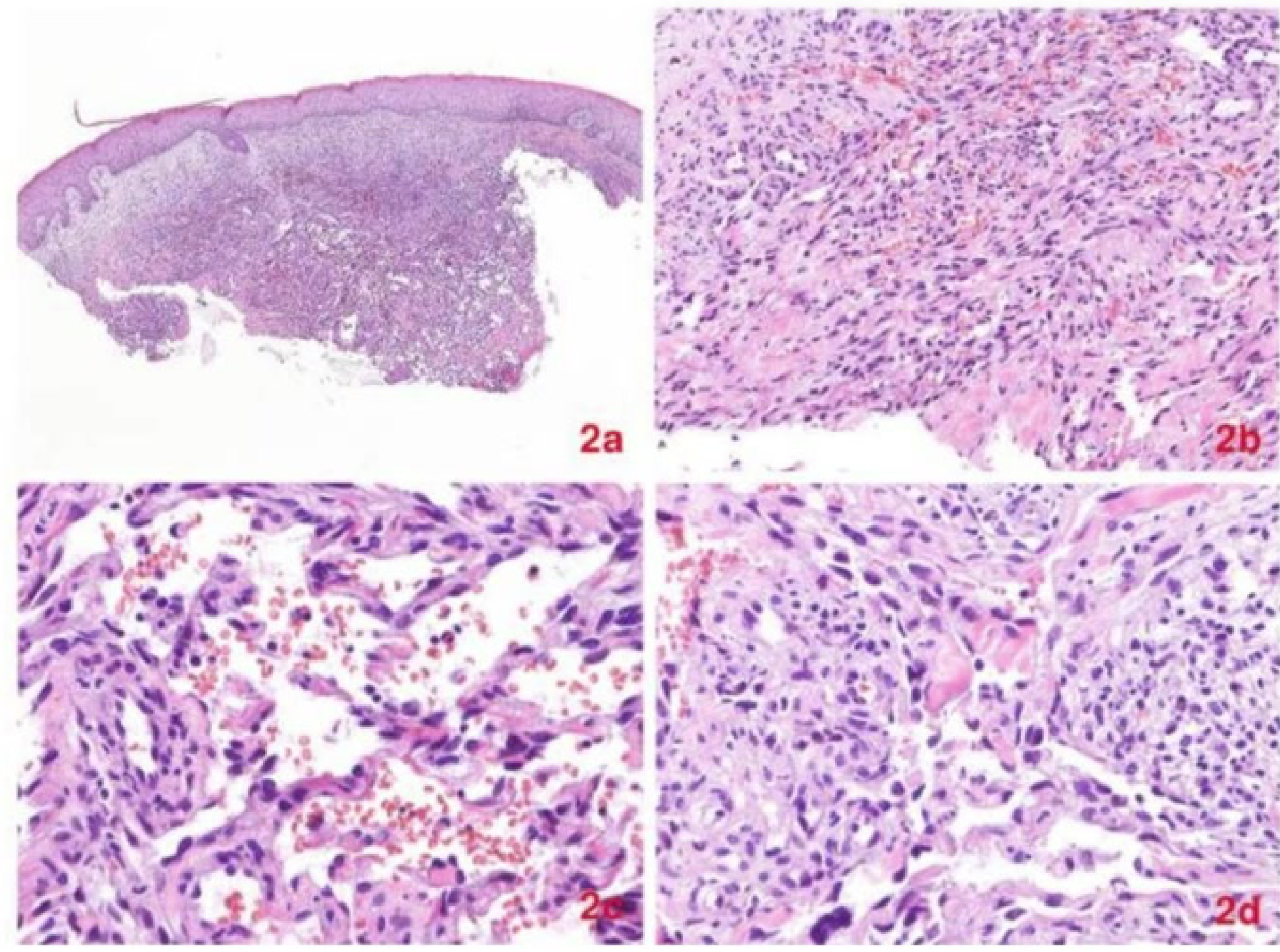
Histopathological Stages52
Early “Patch-Stage” Lesion
- Characterized by the proliferation of small, jagged, endothelial-lined spaces surrounding normal dermal vessels.
- Slit-like vascular spaces dissecting collagen bundles, running parallel to the endothelium.
- Presence of extravasated erythrocytes and lymphocytes.
Advanced Stage
- Accumulation of spindle-shaped cells, which are considered the primary tumor cells of Kaposi Sarcoma.
- Presence of intra- and extracellular hyaline globules.
- Increased mitotic activity.
Diagnosis53
Diagnosis is supported by identifying HHV8 in peripheral blood as well as within the lesional tissue.
Treatment and Prognosis
Management options include:
- Local excision
- Radiation therapy
- Chemotherapy
- Adjustment of immunosuppressive medications
While local recurrence is common in AIDS-KS patients, overall survival is closely linked to the patient’s immunological status. In advanced or aggressive cases, the mortality rate can reach 20–25%.
- Highly aggressive in AIDS patients; treatment often includes HAART (Highly Active Antiretroviral Therapy) to manage the underlying HIV infection.
Conclusion54
Summary
Mesenchymal tumours of the oral cavity encompass a broad spectrum from simple reactive lesions like fibroepithelial polyps to highly aggressive malignancies like rhabdomyosarcoma. Accurate diagnosis relies on a combination of clinical history (especially for vascular lesions), imaging, and definitive histopathology.
Footnotes
-
Original PDF page 1: L14 Soft tissue (mesenchymal) tumours slides, p.1 ↩
-
Original PDF page 2: L14 Soft tissue (mesenchymal) tumours slides, p.2 ↩
-
Original PDF page 3: L14 Soft tissue (mesenchymal) tumours slides, p.3 ↩
-
Original PDF page 4: L14 Soft tissue (mesenchymal) tumours slides, p.4 ↩
-
Original PDF page 5: L14 Soft tissue (mesenchymal) tumours slides, p.5 ↩
-
Original PDF page 6: L14 Soft tissue (mesenchymal) tumours slides, p.6 ↩
-
Original PDF page 7: L14 Soft tissue (mesenchymal) tumours slides, p.7 ↩
-
Original PDF page 8: L14 Soft tissue (mesenchymal) tumours slides, p.8 ↩
-
Original PDF page 9: L14 Soft tissue (mesenchymal) tumours slides, p.9 ↩
-
Original PDF page 10: L14 Soft tissue (mesenchymal) tumours slides, p.10 ↩
-
Original PDF page 11: L14 Soft tissue (mesenchymal) tumours slides, p.11 ↩
-
Original PDF page 12: L14 Soft tissue (mesenchymal) tumours slides, p.12 ↩
-
Original PDF page 13: L14 Soft tissue (mesenchymal) tumours slides, p.13 ↩
-
Original PDF page 14: L14 Soft tissue (mesenchymal) tumours slides, p.14 ↩
-
Original PDF page 15: L14 Soft tissue (mesenchymal) tumours slides, p.15 ↩
-
Original PDF page 16: L14 Soft tissue (mesenchymal) tumours slides, p.16 ↩
-
Original PDF page 17: L14 Soft tissue (mesenchymal) tumours slides, p.17 ↩
-
Original PDF page 18: L14 Soft tissue (mesenchymal) tumours slides, p.18 ↩
-
Original PDF page 19: L14 Soft tissue (mesenchymal) tumours slides, p.19 ↩
-
Original PDF page 20: L14 Soft tissue (mesenchymal) tumours slides, p.20 ↩
-
Original PDF page 21: L14 Soft tissue (mesenchymal) tumours slides, p.21 ↩
-
Original PDF page 22: L14 Soft tissue (mesenchymal) tumours slides, p.22 ↩
-
Original PDF page 23: L14 Soft tissue (mesenchymal) tumours slides, p.23 ↩
-
Original PDF page 24: L14 Soft tissue (mesenchymal) tumours slides, p.24 ↩
-
Original PDF page 25: L14 Soft tissue (mesenchymal) tumours slides, p.25 ↩
-
Original PDF page 26: L14 Soft tissue (mesenchymal) tumours slides, p.26 ↩
-
Original PDF page 27: L14 Soft tissue (mesenchymal) tumours slides, p.27 ↩
-
Original PDF page 28: L14 Soft tissue (mesenchymal) tumours slides, p.28 ↩
-
Original PDF page 29: L14 Soft tissue (mesenchymal) tumours slides, p.29 ↩
-
Original PDF page 30: L14 Soft tissue (mesenchymal) tumours slides, p.30 ↩
-
Original PDF page 31: L14 Soft tissue (mesenchymal) tumours slides, p.31 ↩
-
Original PDF page 32: L14 Soft tissue (mesenchymal) tumours slides, p.32 ↩
-
Original PDF page 33: L14 Soft tissue (mesenchymal) tumours slides, p.33 ↩
-
Original PDF page 34: L14 Soft tissue (mesenchymal) tumours slides, p.34 ↩
-
Original PDF page 35: L14 Soft tissue (mesenchymal) tumours slides, p.35 ↩
-
Original PDF page 36: L14 Soft tissue (mesenchymal) tumours slides, p.36 ↩
-
Original PDF page 37: L14 Soft tissue (mesenchymal) tumours slides, p.37 ↩
-
Original PDF page 38: L14 Soft tissue (mesenchymal) tumours slides, p.38 ↩
-
Original PDF page 39: L14 Soft tissue (mesenchymal) tumours slides, p.39 ↩
-
Original PDF page 40: L14 Soft tissue (mesenchymal) tumours slides, p.40 ↩
-
Original PDF page 41: L14 Soft tissue (mesenchymal) tumours slides, p.41 ↩
-
Original PDF page 42: L14 Soft tissue (mesenchymal) tumours slides, p.42 ↩
-
Original PDF page 43: L14 Soft tissue (mesenchymal) tumours slides, p.43 ↩
-
Original PDF page 44: L14 Soft tissue (mesenchymal) tumours slides, p.44 ↩
-
Original PDF page 45: L14 Soft tissue (mesenchymal) tumours slides, p.45 ↩
-
Original PDF page 46: L14 Soft tissue (mesenchymal) tumours slides, p.46 ↩
-
Original PDF page 47: L14 Soft tissue (mesenchymal) tumours slides, p.47 ↩
-
Original PDF page 48: L14 Soft tissue (mesenchymal) tumours slides, p.48 ↩
-
Original PDF page 49: L14 Soft tissue (mesenchymal) tumours slides, p.49 ↩
-
Original PDF page 50: L14 Soft tissue (mesenchymal) tumours slides, p.50 ↩
-
Original PDF page 51: L14 Soft tissue (mesenchymal) tumours slides, p.51 ↩
-
Original PDF page 52: L14 Soft tissue (mesenchymal) tumours slides, p.52 ↩
-
Original PDF page 53: L14 Soft tissue (mesenchymal) tumours slides, p.53 ↩
-
Original PDF page 54: L14 Soft tissue (mesenchymal) tumours slides, p.54 ↩