Haematological Diseases and Immunodeficiency1
Overview
This lecture provides an overview of red blood cell disorders, bleeding and platelet abnormalities, haematological malignancies, and immunodeficiencies, with a specific focus on their oral manifestations and implications for dental management.
Learning Outcomes2
Upon completion of this section, students should be able to:
- Gain an understanding of an overview of haematological diseases and immunodeficiency.
- Recognise the oral manifestations and management of patients with these conditions.
- Develop an awareness of when medically compromised patients should be referred to secondary care.
- Develop an awareness of when to consult a physician or specialist regarding systemic conditions that may impact dental treatment.
Anaemia
Anaemia is a term used for either a decrease in the volume of red blood cells (RBC) or in the concentration of haemoglobin.
Pathophysiology3
- Results from decreased production of RBCs or increased destruction or loss (trauma, menstruation, GI bleeding) of RBCs.
General Symptoms
Symptoms are related to the reduced oxygen-carrying capacity of the blood:
- Tiredness and shortness of breath
- Tachycardia and palpitations
- Pallor of the mucous membrane
Iron Deficiency Anaemia
Iron-deficiency anaemia is the most common cause of anaemia and is classified as hypochromic microcytic anaemia.
Causes4
- Decreased dietary intake of iron
- Decreased absorption of iron (e.g., Coeliac disease)
- Excessive blood loss (e.g., menstrual or GI bleeding)
- Increased demand for red blood cells (e.g., pregnancy)
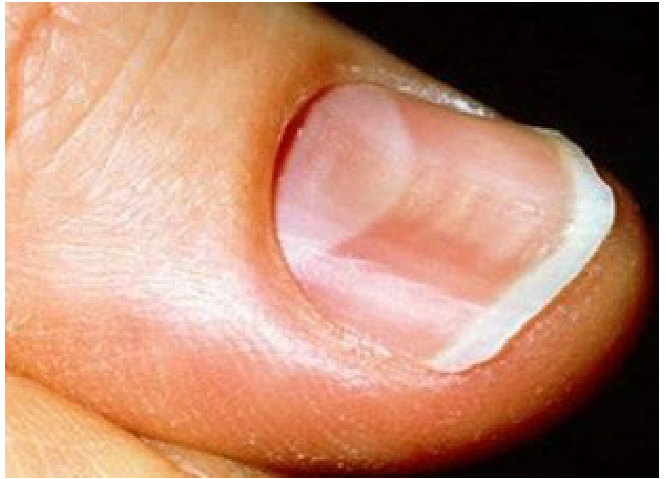 |  |
Signs and Symptoms
- Fatigue and dyspnoea
- Koilonychia (spoon-shaped nails)
- Pica (craving for specific foods/dirt)
- Blue sclera
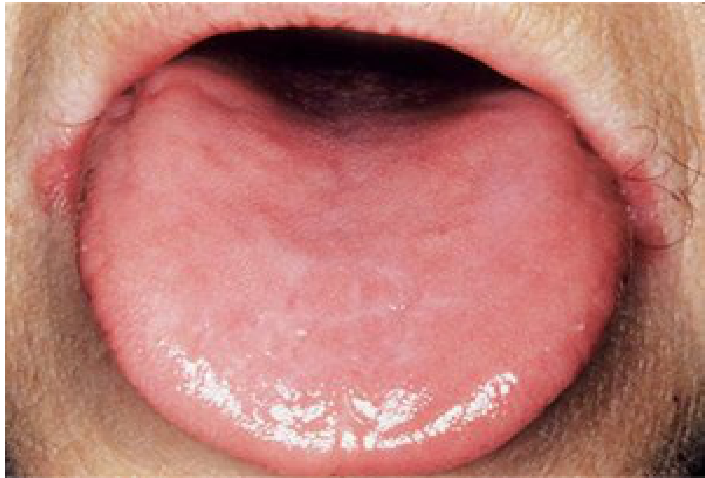
Oral Findings
- Pale oral mucosa
- Depapillated atrophic tongue
- Glossodynia (burning sensation)
- Candidiasis and angular cheilitis
- Aphthous-like ulcers
Plummer-Vinson Syndrome
Plummer-Vinson syndrome (also known as Patterson-Brown-Kelly syndrome) is a severe and chronic form of iron deficiency.
Clinical Characteristics
- Typically affects middle-aged females.
- Atrophic glossitis: Smooth, atrophic, and red tongue.
- Pharyngeal and oesophageal webbing: Leads to dysphagia.
- Malignancy Risk: Predisposition to post-cricoid and oral squamous cell carcinoma.
Dental Management Considerations for Anaemia
Anaesthesia5
- In poorly controlled anaemia: Use local anaesthesia (LA) without epinephrine, as it may aggravate cardiac symptoms.
Anxiety and Sedation
- Nitrous oxide can be used.
- General Anaesthesia (GA) should be avoided if the Hb level is below 10g/dL.
Medical Referral
- Iron deficiency in men may indicate an occult GI cancer; refer for further evaluation.
Diagnosis of Iron Deficiency
Diagnosis involves a Full Blood Count (FBC): reduced Hb, reduced Mean Corpuscular Volume (MCV), low serum iron/ferritin, and high Total Iron Binding Capacity (TIBC).
Thalassemia
Thalassemia is a hereditary haemoglobinopathy characterised by a reduction in the production of globin chains in the haemoglobin molecule.
Epidemiology and Classification6
- More prevalent in Mediterranean, African, and Asian populations.
- Types:
- Alpha-thalassemia
- Beta-thalassemia
Pathogenesis
- Caused by deletions or mutations in the alpha or beta globin genes.
- Results in decreased haemoglobin production and the formation of malformed RBCs.
- Severity increases with the number of defective genes.
Systemic Features7
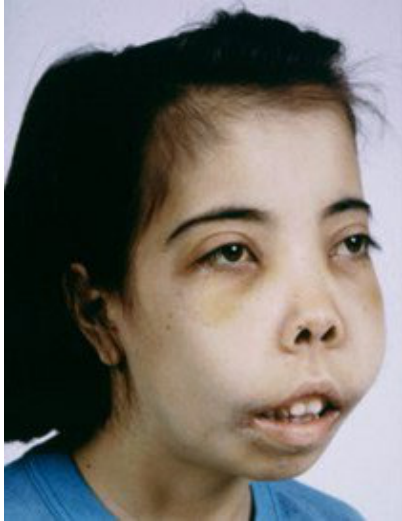
- Severe jaundice, pallor, growth retardation, and splenomegaly (typically appearing in the 1st year of life).
- Typical symptoms of anaemia.
 |  |
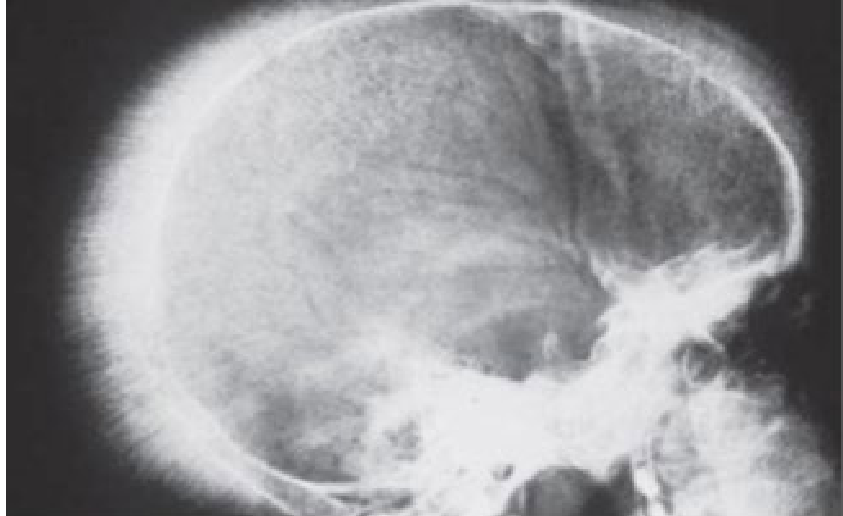 |
Skeletal and Radiographic Findings
Bone expansion occurs as a result of intramedullary haematopoiesis:
- Maxillary protrusion with spacing of teeth.
- Thin cortical plates and spongy marrow.
- Thin, widely spaced trabeculae.
- “Hair-on-end” appearance on a lateral skull radiograph.
Sickle Cell Anaemia
Sickle cell anaemia is a hereditary haemoglobinopathy in which red cells contain an abnormal haemoglobin, HbS.
Pathogenesis8
- Autosomal recessive disorder.
- More common in Mediterranean, African, and Far East countries.
- Caused by a point mutation of the beta-globin gene.
- Red cells become rigid and curved (sickle-shaped).
- Deformed sickle cells are highly vulnerable to haemolysis.
- Newborn babies are screened for sickle cell status.
- Deformed cells can block small vessels.
- Diagnosis: Sickle cell solubility test and haemoglobin electrophoresis.
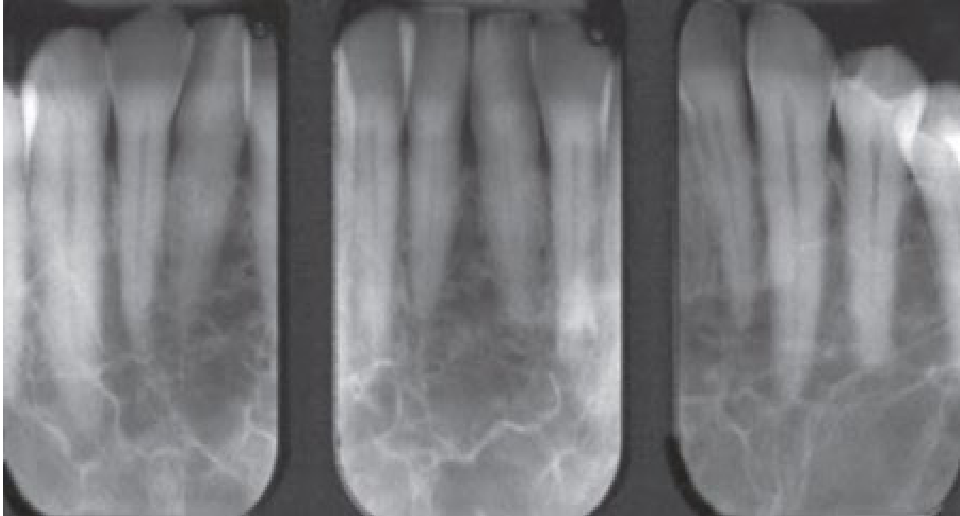
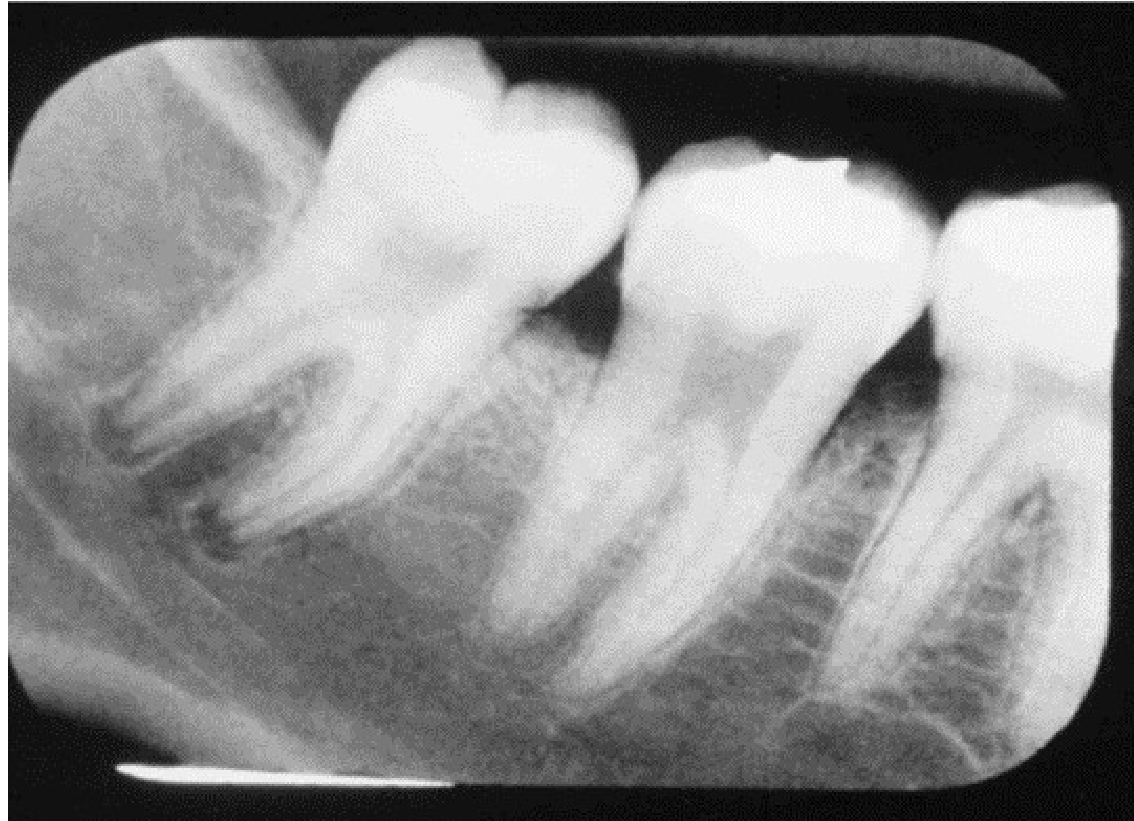
Clinical and Radiographic Findings9
- Pallor or jaundice of the oral mucosa.
- Widely spaced trabeculae.
- Step-ladder appearance: Characteristic bone trabeculation seen in periapical (PA) x-rays.
- Mandibular bone pain and osteomyelitis.
- Asymptomatic pulpal necrosis.
- Maxillary protrusion.
- Delayed eruption and dental hypoplasia.
Bleeding and Platelet Disorders
Info
Hemostasis involves vasoconstriction, platelet plug formation, and activation of the coagulation pathway to form a fibrin clot.
Vascular Disorders10
- Capillary fragility.
- e.g., Vitamin C deficiency/Scurvy.
- Hereditary haemorrhagic telangiectasia.
- Also known as Osler-Weber-Rendu Syndrome.
Platelet Disorders
- Abnormal platelet number or function.
- Quantitative: Thrombocytopenia (low count)
- Qualitative: Thrombocytopathy (abnormal function, e.g., Aspirin use, Von Willebrand disease).
- Quantitative: Thrombocytopenia (low count)
- Inability to develop a temporary clot.
Coagulation Disorders
- Defects in coagulation leading to an inability to form a definitive clot.
- Inherited: e.g., von Willebrand’s disease, haemophilia.
- Acquired: e.g., liver disease, anticoagulants.
Fibrolytic Disorders
- Inability to destroy free plasmin.
Assessing Risk for Bleeding
Patient History11
- Bleeding problems in relatives (e.g., von Willebrand’s disease, haemophilia).
- History of bleeding after surgery or tooth extractions.
- Bleeding after trauma (cuts).
- Spontaneous bleeding from the nose, mouth, or GI tract.
Medications and Treatments
- Antiplatelets.
- Long-term antibiotic therapy.
- Some herbal preparations.
- e.g., Ginkgo biloba, Garlic.
- Chemotherapy.
Associated Medical Conditions
- Leukaemia and thrombocytopenia.
- Advanced liver disease.
- Renal failure.
Thrombocytopenia
Thrombocytopenia is defined as low platelet levels in the blood. Clinical evidence is typically not visible until the platelet level falls below 100,000/µl (Normal range: 150,000 to 450,000/µl).
Causes of Decreased Production12
- Bone marrow dysfunction or infiltration by malignant cells (leukaemia, myeloma, bone metastasis).
- Toxic effects of chemotherapeutic drugs.
- Liver disease.
Causes of Increased Consumption or Destruction
- Drug-induced immunologic responses (heparin, quinidine, methyldopa).
- Systemic diseases: SLE and HIV infection.
- Vaccinations and viral infections.
- Immune (idiopathic) thrombocytopenic purpura (ITP).
Other Causes
- Increased splenic sequestration (portal hypertension due to liver disease, tumour infiltration).
Oral Manifestations13
- Petechiae and ecchymosis.
- Spontaneous gingival bleeding.
- Postoperative bleeding.
Dental Management Considerations
- Obtain a thorough history, clinical exam, and relevant tests (platelet count).
- Refer and consult a haematologist if necessary.
- Use local measures to control bleeding.
- Contraindication: Do not prescribe aspirin.
- Be aware of the risk of infection in patients with bone marrow suppression (e.g., leukaemia).
- Guidelines for Platelet Counts:
- Count > 50,000/µL: Extractions and minor surgery possible
- Count > 80,000/µL: Major oral surgery possible
- Count < 30,000/µL: Only non-surgical procedures
- Use Acetaminophen for pain management instead of Aspirin/NSAIDs.
Local Haemostatic Agents
Physical and Mechanical Agents14
- Gauze: 2″×2″ sterile pads; applied with pressure (closing or finger pressure).
- Surgicel®: Oxidized regenerated cellulose; exerts a physical effect by swelling on contact with blood; becomes gelatinous after 24-48 hours.
- Gelfoam®: Absorbable gelatin sponge.
- Atraumatic Technique: Minimally traumatic surgery and precise suturing.

Physiological and Chemical Agents
- Avitene®: Microfibrillar collagen hemostat; attracts platelets and triggers aggregation.
- Thrombostat®: Topical thrombin; converts fibrinogen to fibrin.
Antifibrinolytic Agents
- Tranexamic acid: Competitive inhibitor of plasminogen activation (available as 4.8% mouthwash in Europe; tablets or injections in the USA).
- ε-Aminocaproic acid (EACA/Amicar®): Competitive inhibitor of plasminogen activation; used as a rinse (25% syrup).
Platelet Function Disorders
Inherited Disorders15
- Von Willebrand’s disease: May also involve a secondary factor VIII deficiency.
Acquired Platelet Dysfunction
- Drug-induced: Aspirin, clopidogrel, NSAIDs, beta-lactam antibiotics (penicillin), calcium channel blockers, and phenytoin.
- End-stage renal disease: Uraemia (high urea levels) can result in circulating toxins that impair platelet function.
- Alcohol ingestion.
Von Willebrand Disease
Von Willebrand disease is the most common inherited bleeding disorder, caused by an inherited gene mutation on chromosome 12 affecting both males and females.
Pathophysiology
- Characterised by deficient or defective von Willebrand factor (vWF).
- vWF serves as a carrier protein for Factor VIII.
- Patients experience both platelet dysfunction and Factor VIII deficiency.
Clinical Features
- Common findings: Ecchymoses and nose bleeds.
- Prolonged bleeding after tooth extraction.
- Severe forms may present with haemarthroses.
Coagulation Disorders
Hereditary Disorders16
- Von Willebrand’s disease
- Haemophilia:
- Haemophilia A (Factor VIII deficiency)
- Haemophilia B (Factor IX deficiency)
Acquired Disorders
- Anticoagulants: Warfarin and Heparin therapy.
- Liver Disease: Viral hepatitis and alcoholism.
- Vitamin K Deficiency: Caused by malabsorption or long-term use of broad-spectrum antibiotics.
Haemophilia A
Haemophilia A is an X-linked recessive disorder characterized by factor VIII deficiency. It primarily affects males, while females are usually carriers. Normal haemostasis requires at least 30% factor VIII activity.
Severity Classification17
- Mild haemophilia: Factor VIII level between 5-30%; patients are often asymptomatic
- Often asymptomatic until trauma or surgery..
- Moderate haemophilia: Factor VIII level between 1-5%.
- Severe haemophilia: Factor VIII level less than 1
- Characterized by spontaneous bleeding.%.
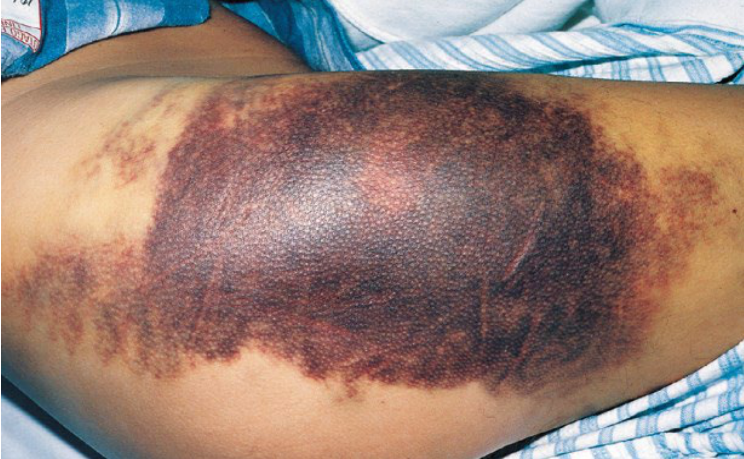
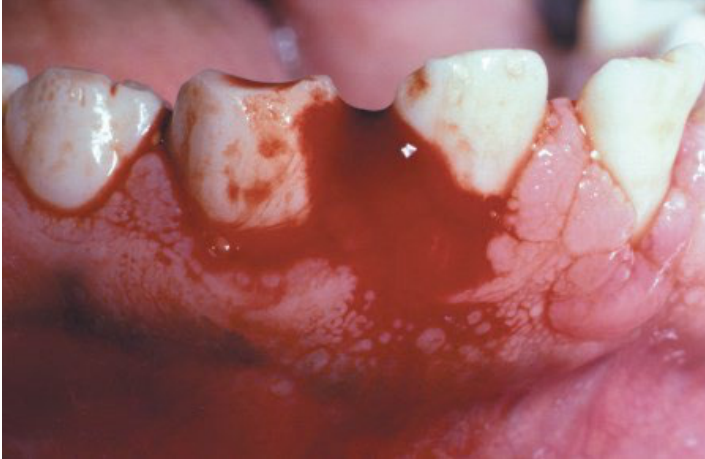
General and Oral Findings18
- Common findings: Ecchymoses, hemarthrosis, and dissecting haematomas.
- Persistent bleeding after dental surgery.
- Bleeding after tooth extraction (may be the first evidence of mild haemophilia).
- Formation of liver clots and haematomas.
- Spontaneous mucosal or gingival bleeding.
- Hemarthrosis of the TMJ (rare finding).
 |  |
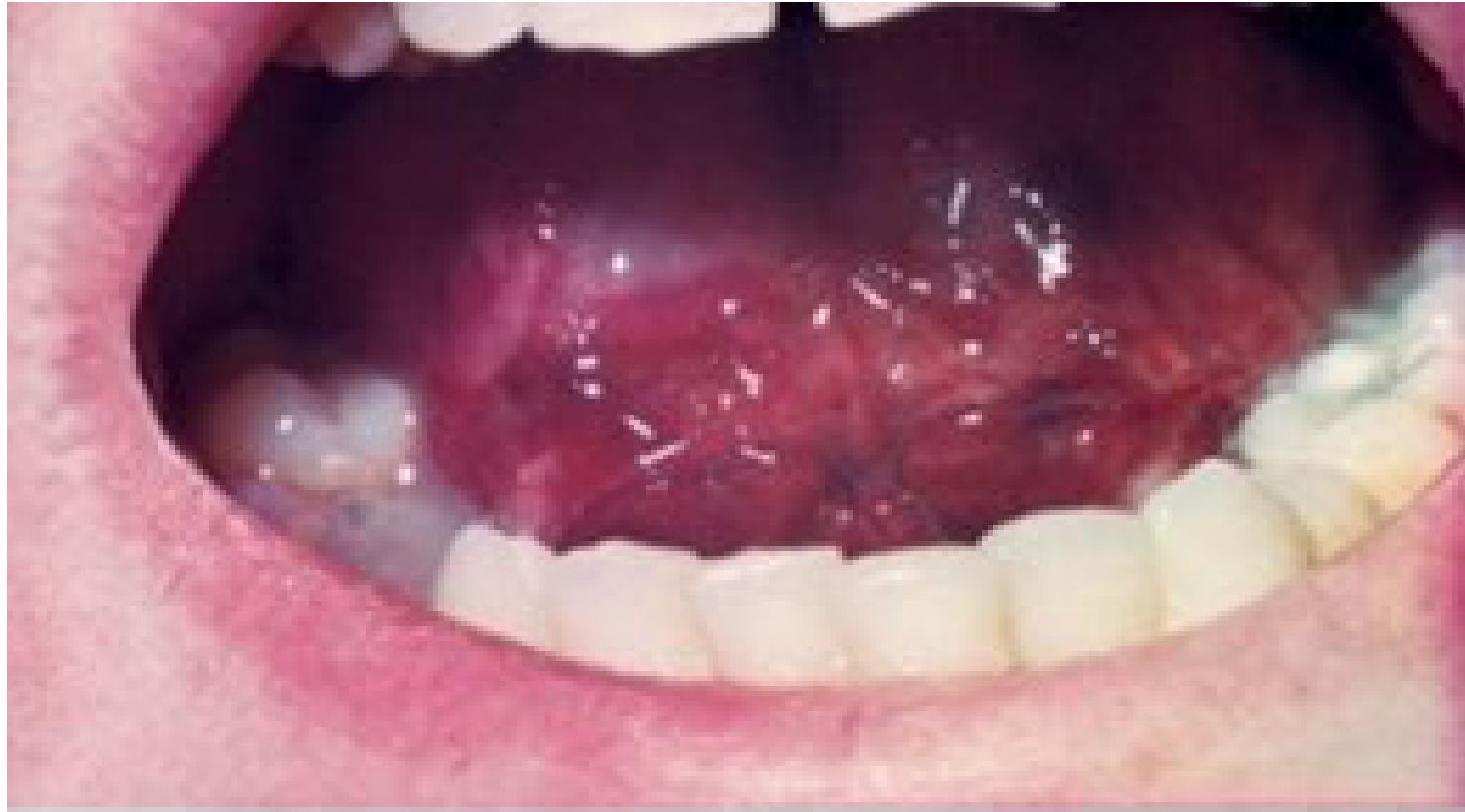 |
Haemophilia B
Haemophilia B, also known as Christmas disease, is a sex-linked recessive disorder characterised by Factor IX deficiency.
Clinical Profile19
- Clinical features are identical to Haemophilia A.
- Inherited as an X-linked recessive trait.
- Classified as mild, moderate, or severe (20-45% of cases are severe).
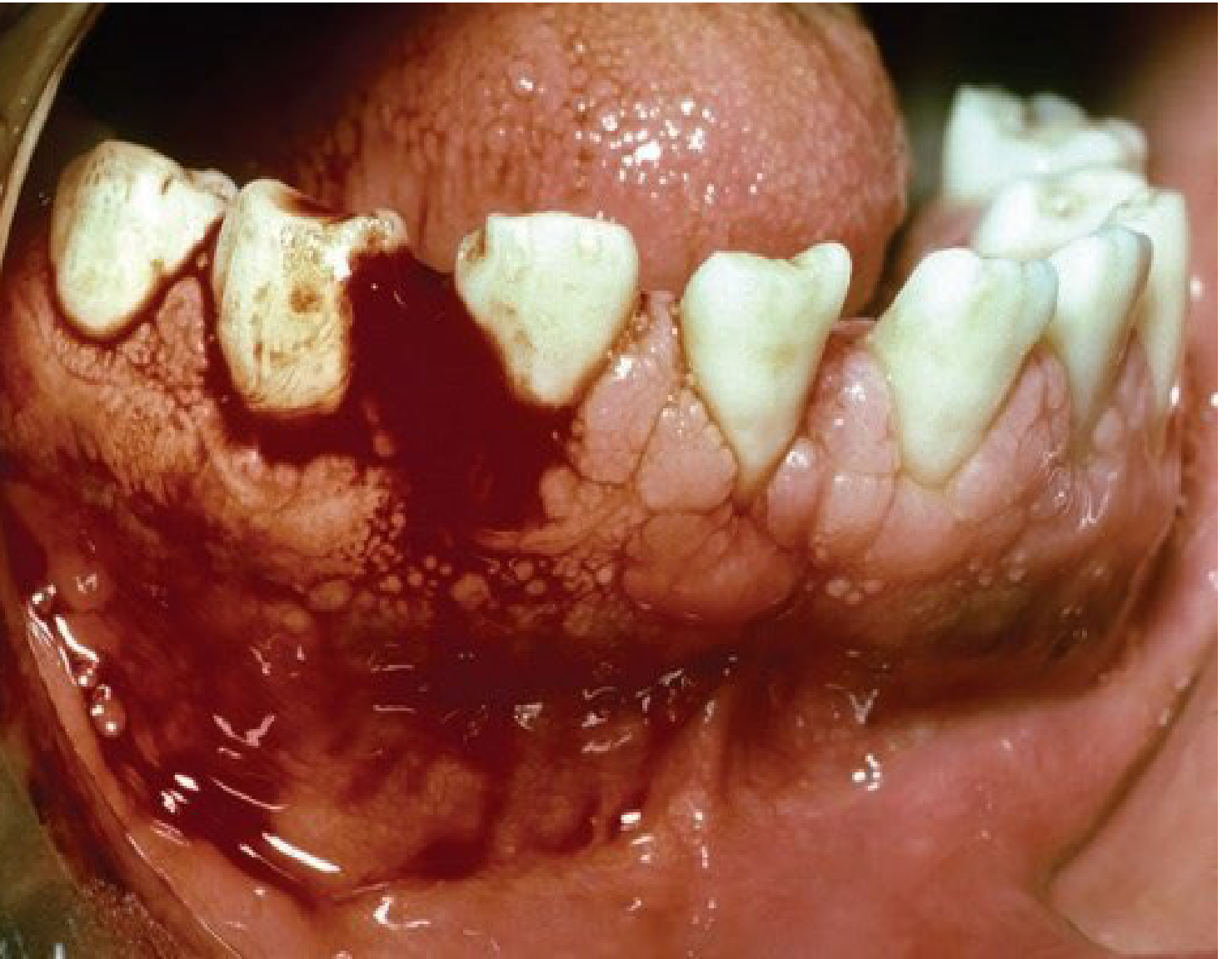
Diagnosis and Management
- Lab tests: Prolonged PTT (partial thromboplastin time) and confirmed Factor IX deficiency.
- Medical management: Factor IX replacement therapy.
Dental Management for Patients on Warfarin
Pre-operative Assessment20
- Determine the reason for anticoagulant therapy.
- Confirm the INR level, ideally within 72 hours of the scheduled procedure.
- Consult with the patient’s physician if necessary.
Clinical Guidelines based on INR
- INR ≤ 3.5: Minor procedures can be performed using local haemostatic measures.
- INR > 3.5: Consult the physician; delay the procedure by 3-5 days and re-confirm the INR level
- Physician consultation may involve adjusting the dosage..
- Major Oral Surgery: INR should be less than 3.0.
Emergency Measures
- If post-operative bleeding is uncontrollable, Vitamin K or fresh frozen plasma may be used.
Haematological Malignancies
Leukaemia
Leukaemia is a cancer of the white blood cells (WBCs) that affects the bone marrow and circulating blood, involving the exponential proliferation of clonal or lymphoid cells.
Classification21
- Acute Leukaemia: Sudden onset and rapid course; involves immature, undifferentiated WBCs.
- Chronic Leukaemia: Slower onset; involves more mature, functional cells.
- Types: ALL, AML, CLL, CML.
Clinical Signs22
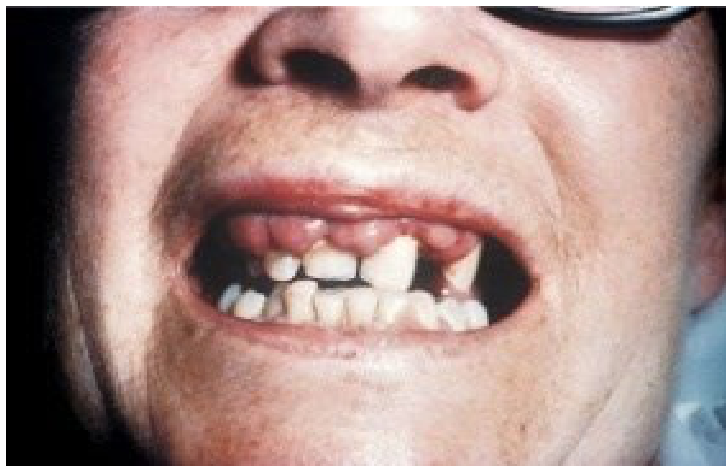
- Mucosal pallor (important sign in children).
- Gingival bleeding, petechiae, or purpura.
- Spontaneous gingival bleeding (a key diagnostic sign)
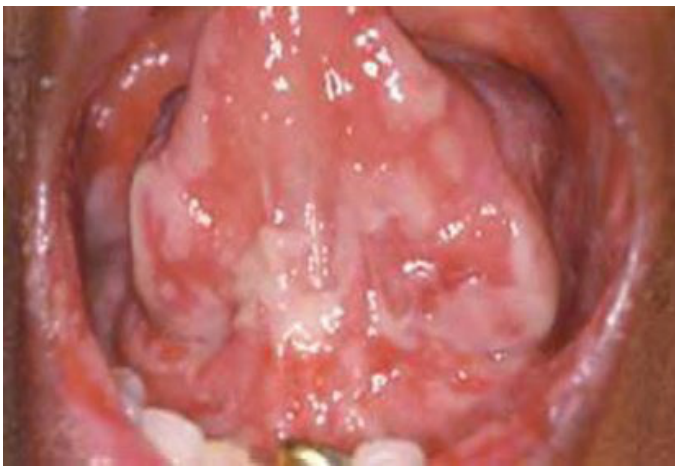
- Gingival enlargement (soft, boggy, leukemic infiltrates)
- Oral ulcers (herpetic, neutropenic, or drug-induced).
- Oral infections (e.g., candidiasis, CMV).
- Cervical lymphadenopathy.
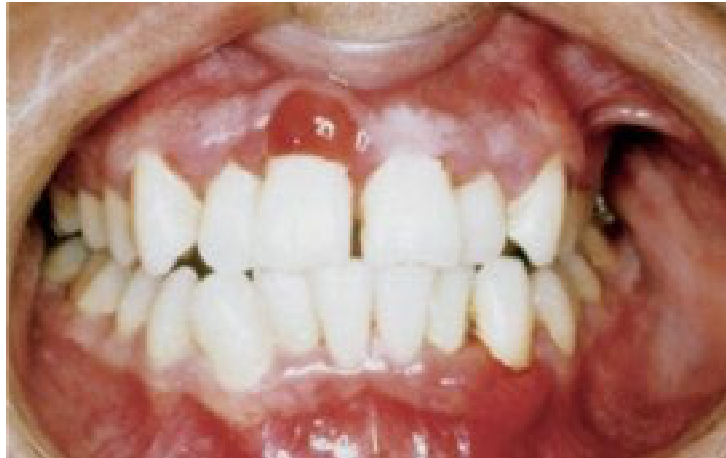 | 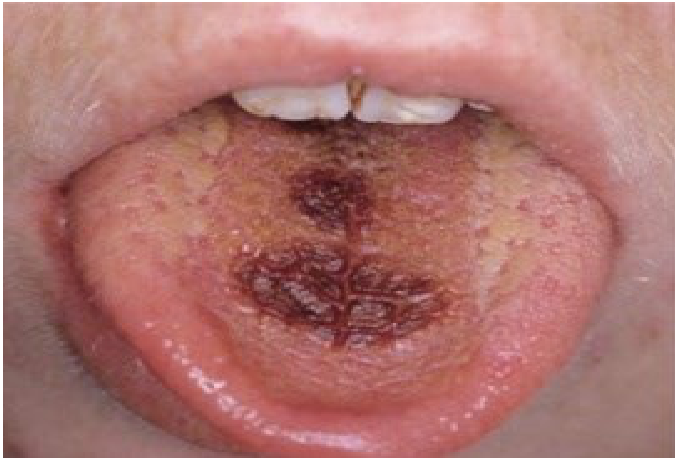 |
 |  |
Specific Pathologies
- Gingival Enlargement: Localised or generalised; caused by leukemic infiltrates. Gingiva appears boggy and bleeds easily.
- Myeloid Sarcoma: A localised mass of leukemic cells that can involve the maxilla, palate, gingiva, tongue, or oral mucosa.
Lymphoma
Hodgkin Lymphoma
Hodgkin Lymphoma (HL) is a B lymphocyte neoplasm that presents as discrete masses in lymphoid organs.
Pathophysiology and Epidemiology
- Contains the characteristic Reed-Sternberg cell (large, bi-nucleated tumour cell
- Reed-Sternberg cells often have a characteristic "owl-eye" appearance).
- Most common in adolescents and young adults (bimodal incidence: 20-30 and 50-70 years).
- EBV is linked to 40% of cases.
Clinical Presentation
- Typically manifests as painless, firm, enlarged lymph nodes with a rubbery consistency (commonly mediastinal and cervical).
- Oral involvement is rare.
- Systemic symptoms: Pruritus, fatigue, fever, weight loss, and night sweats.
Non-Hodgkin Lymphoma
Non-Hodgkin lymphoma (NHL) is a large group of lymphoproliferative disorders derived from B, T, and natural killer cells (B-cell proliferation accounts for 85-90% of cases).
Risk Factors and Age
- Can occur at any age (median age at diagnosis is 67 years).
- Causative agents: Genetic factors, infectious agents, autoimmune diseases (e.g., Sjögren syndrome), and immunodeficiency states (e.g., AIDS).
Risk Factor: Sjögren Syndrome
Patients with long-term Sjögren’s Syndrome have a significantly higher risk of developing Non-Hodgkin Lymphoma.
Clinical Presentation
- Involvement can be nodal (painless, firm enlarged cervical lymph nodes) or extra-nodal.
- Oral Findings:
- Oral submucosal mass (may or may not be ulcerated).
- Involvement of salivary glands (especially those affected by Sjögren syndrome).
- Jaw bone involvement in African children with Burkitt’s Lymphoma.
Multiple Myeloma
Multiple myeloma is a malignant neoplasm composed of plasma cells, occurring as a disseminated disease involving many bones.
Clinical Features
- Typically affects men over 50 years; presents with persistent bone pain.
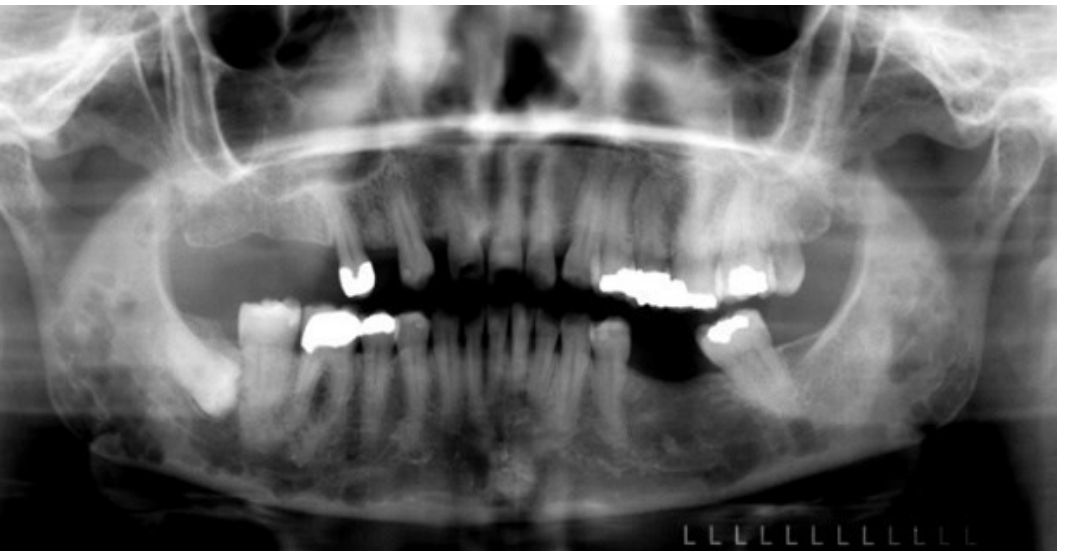
- Skeletal Involvement: Multiple osteolytic “punched-out” lesions or mottled areas on radiographs. Commonly affects vertebrae, sternum, ribs, and pelvic bones
- "Punched-out" radiolucencies are especially prominent on the skull.
- Jaw Involvement: Osteolytic lesions of the jaws occur in 30% of patients.
- Systemic Complications:
- Serum hypercalcaemia and potential pathological fractures.
- Fever and recurrent infections due to neutropenia.
- Proteinuria and renal failure.
- Amyloid deposition in various tissues (heart, liver, nervous tissue).
Radiographic and Oral Findings23
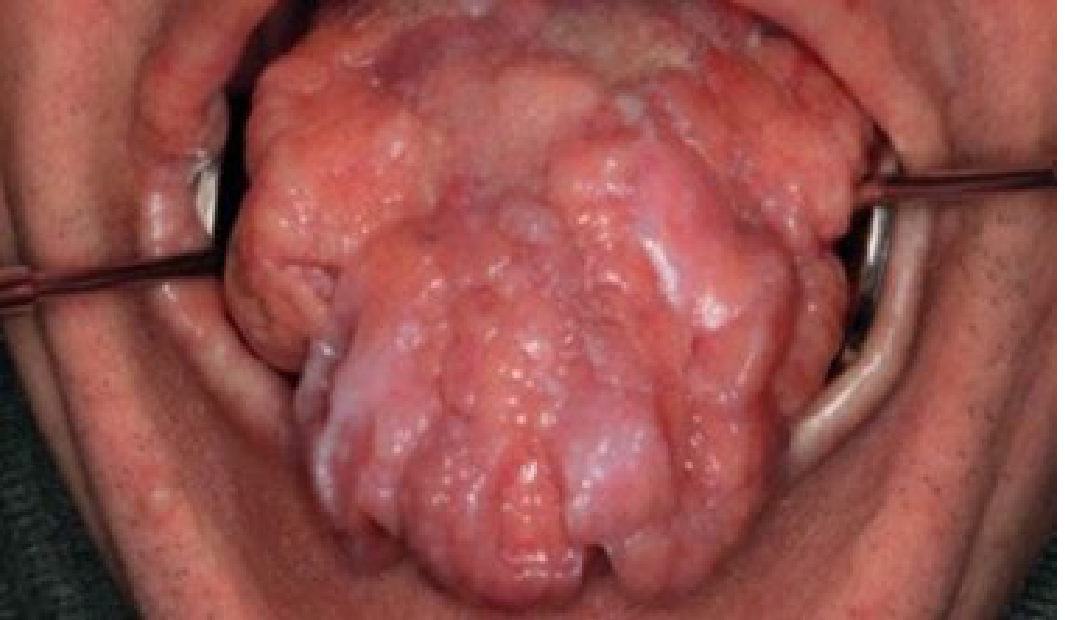
- “Punched-out” lesions: May cause bone pain, paraesthesia, and cortical enlargement; can be the initial manifestation of the disease.
- Macroglossia: Caused by amyloid deposition in the tongue, leading to pain.
- Haematological signs: Petechiae, pallor, and increased susceptibility to bleeding and infections.
 |  |
Treatment Complications
-
MRONJ (Medication-related osteonecrosis of the jaw): A potentially serious complication of long-term bisphosphonate therapy, commonly observed in the mandible.
-
Distinction: Unlike Candida, Hairy Leukoplakia cannot be wiped off.
Immunodeficiency24
Immunodeficiency refers to a state in which the immune system’s ability to fight infectious diseases and cancer is compromised or entirely absent.
Primary Immunodeficiency
Characteristics of Primary Immunodeficiency25
- Genetically based.
- Predominantly involves B-cell or T-cell defects, a combination of both, or selective immunodeficiencies (e.g., IgA deficiency).
- Results in life-threatening conditions.
- Affected individuals tend to die young as a result of recurrent infection.
Clinical Manifestations
- Candida infections are often predominant in these patients.
- Patients are more susceptible to periodontal infections.
Secondary Immunodeficiency26
Secondary immunodeficiency can arise from various underlying conditions and external factors:
- Malignancies
- Leukaemia, lymphoma, multiple myeloma
- Medications
- Corticosteroids and immunosuppressants
- Chemotherapy
- Health and Lifestyle Factors
- Malnutrition
- Autoimmune disorders
- Diabetes mellitus
- Chronic renal failure
- Environmental and Infectious Factors
- Radiation
- Infections (e.g., HIV, TB)
Human Immunodeficiency Virus Infection
Pathophysiology and Transmission27
- Caused by an RNA retrovirus infection.
- Spread through:
- Sexual contact
- Parenteral exposure to blood
- Mother to foetus
- Immune deficiency is due to damage to CD4 T-lymphocytes.
Predisposition to Complications
Infected individuals are predisposed to:
- Viruses and virally induced malignancies
- Fungi
- Mycobacteria
- Autoimmune disease
- Neurological damage
Clinical Stages and Classification
Clinical Progression Stages
-
Stage I: Seroconversion Illness
- Occurs within 2-3 weeks.
- Symptoms: Fever, malaise, lymphadenopathy, pharyngitis, mouth ulcers.
- Antibodies typically develop within 3-6 months.
-
Stage II: Asymptomatic Stage
- Chronic phase lasting approximately 10 years.
-
Stage III: Persistent Generalized Lymphadenopathy
- Lasts approximately 10 years.
-
Stage IV (A-E): Symptomatic Stage
- Diseases involving the entire body.
- Includes neurological disease, secondary infectious disease, secondary cancers, and other conditions.
Classification of Oral Lesions
- Group I: Lesions strongly associated with HIV infections
- Example: HIV-associated candidosis
- Group II: Lesions less commonly associated with HIV infection
- Example: HIV-associated CMV ulcer
- Group III: Lesions possibly associated with HIV infection
- Example: HIV-associated wart
Oral Manifestations of HIV28
Common oral manifestations observed in HIV disease include:
- Candidiasis
- Hairy leukoplakia
- Kaposi’s sarcoma
- Gingival and periodontal disease
- Ulcers
- Other orofacial conditions

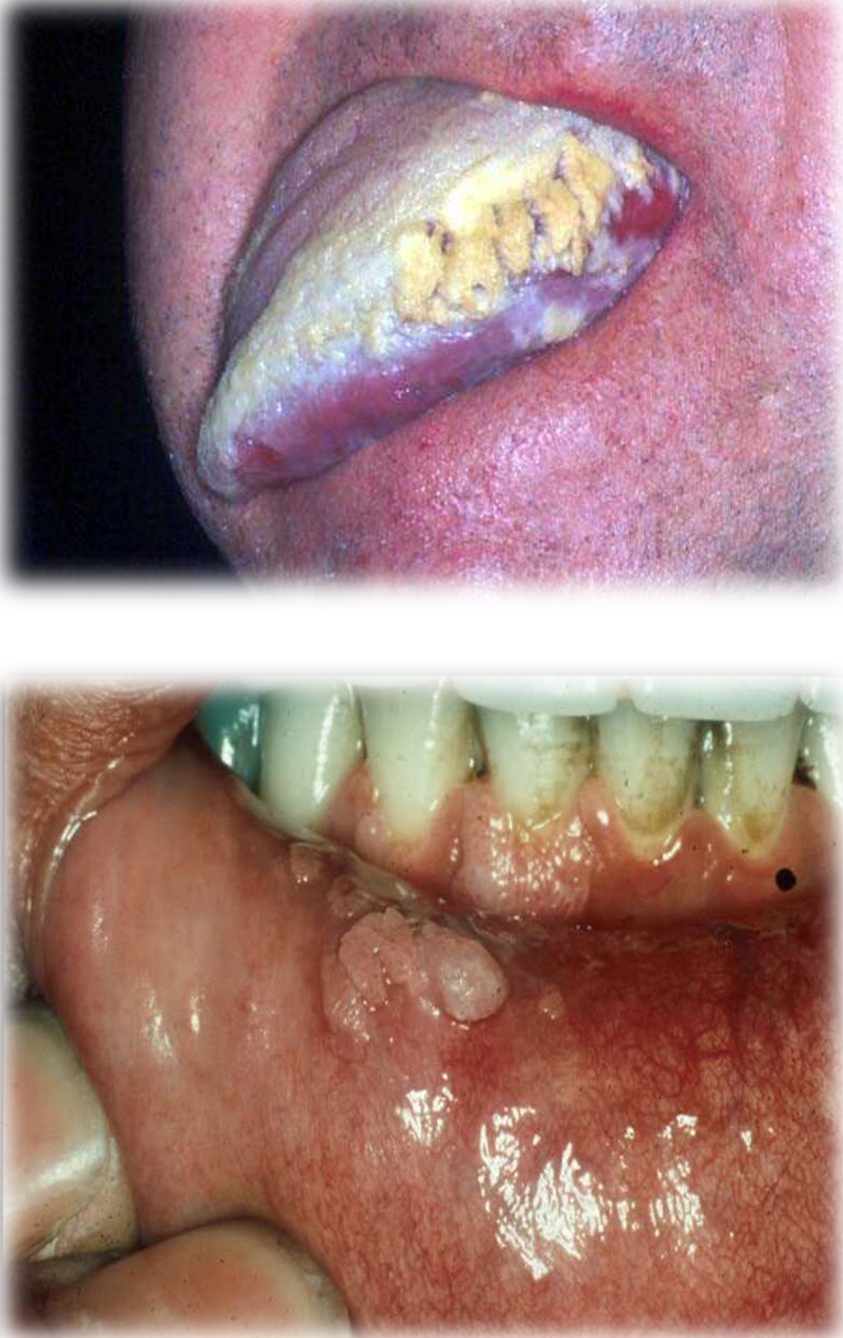
Hairy Leukoplakia
Clinical Features
- Presents as a white patch which cannot be removed.
- Characterized by vertical white folds on the lateral aspects of the tongue.
- Associated with Epstein-Barr Virus (EBV).
Additional Characteristics
- The lesion is not premalignant.
- Usually superimposed by Candida.
- Common in patients with late-stage HIV infection.
- Development may herald the onset of AIDS.
Diagnostic Information
- Diagnosis: Primarily clinical.
- Biopsy: Shows ballooned cells with perinuclear vacuoles; swollen cells contain EBV.
Kaposi’s Sarcoma
Overview
- The most common malignancy in HIV patients.
- Caused by Human Herpesvirus type 8 (HHV-8).
- Arises from vascular endothelial cells.
- Common in homosexuals, though it can occur in all risk groups.
Clinical Presentation
- Involves skin and mucosal surfaces.
- The tip of the nose is a frequent facial site.
- Common oral sites include the hard palate and gingiva.
- Presents as reddish-purple patches that may become nodular and ulcerate.
Diagnosis
- Clinical suspicion must be supported by biopsy.
HIV-Associated Periodontal Diseases
Linear Gingival Erythema
- Characterized by a red band involving the free gingival margins.
- Not related to the accumulation of dental plaque.
Necrotizing Conditions
- Necrotizing Ulcerative Gingivitis: Features punched-out ulceration of the interdental papillae.
- Destructive Periodontitis: Involves necrosis of gingival and periodontal tissues, sometimes resulting in exposure and sequestration of alveolar bone.
- Necrotizing Ulcerative Periodontitis (NUP) is characterized by rapid bone loss.
HIV-Associated Lymphoma
Clinical Presentation
- Oral lesions present as soft tissue enlargement or ulcerative lesions of the gingiva.
- Presents as large, ulcerated masses on the gingiva or palate.
Pathological Associations
- Increased incidence of Non-Hodgkin Lymphoma (NHL).
- Some cases are associated with Epstein-Barr Virus (EBV).
Acquired Immunodeficiency Syndrome29
- AIDS is the final stage of HIV disease.
- Clinical Markers:
- CD4 cell count: <200 cells/mm³
- CD4 cell %: <14%
- Highly active antiretroviral therapy (HAART) has significantly improved the quality and length of survival for many patients.
Diagnosis and Key Points
Clinical and Laboratory Diagnosis
- Diagnosis may be clear when a patient presents with obvious lesions such as Hairy Leukoplakia (HL), candidiasis, or Kaposi’s sarcoma.
- Serological Confirmation: Necessary via Enzyme-Linked Immunosorbent Assay (ELISA).
- Confirmatory Testing: HIV P24 antibody confirmed by Western Blot.
- Viral Detection: Polymerase Chain Reaction (PCR) is used to detect HIV RNA.
Summary of HIV Infection
- Caused by a retrovirus.
- Transmitted sexually, through IV drug abuse, and via blood & blood products.
- Characterized by progressive deterioration of cell-mediated immunity.
- Oral signs and symptoms may be the initial manifestation of the disease.
- Oral candidiasis is the most prevalent oral lesion.
- Hairy leukoplakia may indicate progression to AIDS.
- Kaposi’s sarcoma and lymphomas often occur in the oral regions.
- Associated with neurological and psychological disorders.
- Death is mainly due to opportunistic infections.
Dental Management of HIV Patients
Responsibilities and Clinical Guidelines
- Referral: Refer to a specialist if a diagnosis has not yet been made.
- Knowledge: Understand the disease progression and management strategies.
- Clinical Care:
- Provide oral health care, specifically to avoid
- Avoid aggressive surgery if the patient is severely immunocompromised. causes of infection or pain.
- Avoid needle stick accidents.
- Avoid surgery where possible.
- Treat xerostomia.
- Provide oral health care, specifically to avoid
- Patient Education: Provide oral health education to the patient.
- Ethics and Safety:
- Treat the diagnosis with strict confidentiality.
- Avoid drug interactions with antiretroviral agents.
- Maintain strict adherence to infection control measures.
Footnotes
-
Original PDF page 1: L21 haematological disease and immunodeficiency, p.1 ↩
-
Original PDF page 2: L21 haematological disease and immunodeficiency, p.2 ↩
-
Original PDF page 3: L21 haematological disease and immunodeficiency, p.3 ↩
-
Original PDF page 4: L21 haematological disease and immunodeficiency, p.4 ↩
-
Original PDF page 6: L21 haematological disease and immunodeficiency, p.6 ↩
-
Original PDF page 7: L21 haematological disease and immunodeficiency, p.7 ↩
-
Original PDF page 8: L21 haematological disease and immunodeficiency, p.8 ↩
-
Original PDF page 9: L21 haematological disease and immunodeficiency, p.9 ↩
-
Original PDF page 10: L21 haematological disease and immunodeficiency, p.10 ↩
-
Original PDF page 11: L21 haematological disease and immunodeficiency, p.11 ↩
-
Original PDF page 12: L21 haematological disease and immunodeficiency, p.12 ↩
-
Original PDF page 13: L21 haematological disease and immunodeficiency, p.13 ↩
-
Original PDF page 14: L21 haematological disease and immunodeficiency, p.14 ↩
-
Original PDF page 15: L21 haematological disease and immunodeficiency, p.15 ↩
-
Original PDF page 16: L21 haematological disease and immunodeficiency, p.16 ↩
-
Original PDF page 18: L21 haematological disease and immunodeficiency, p.18 ↩
-
Original PDF page 19: L21 haematological disease and immunodeficiency, p.19 ↩
-
Original PDF page 20: L21 haematological disease and immunodeficiency, p.20 ↩
-
Original PDF page 21: L21 haematological disease and immunodeficiency, p.21 ↩
-
Original PDF page 22: L21 haematological disease and immunodeficiency, p.22 ↩
-
Original PDF page 23: L21 haematological disease and immunodeficiency, p.23 ↩
-
Original PDF page 24: L21 haematological disease and immunodeficiency, p.24 ↩
-
Original PDF page 28: L21 haematological disease and immunodeficiency, p.28 ↩
-
Original PDF page 29: L21 haematological disease and immunodeficiency, p.29 ↩
-
Original PDF page 30: L21 haematological disease and immunodeficiency, p.30 ↩
-
Original PDF page 31: L21 haematological disease and immunodeficiency, p.31 ↩
-
Original PDF page 32: L21 haematological disease and immunodeficiency, p.32 ↩
-
Original PDF page 35: L21 haematological disease and immunodeficiency, p.35 ↩
-
Original PDF page 40: L21 haematological disease and immunodeficiency, p.40 ↩